
Abstract
Chimeric antigen receptor (CAR) T-cell immunotherapy still faces many challenges in the treatment of solid tumors, one of which is T-cell dysfunction or exhaustion. Immunomodulator lenalidomide may improve CAR T-cell function. In this study, the effects of lenalidomide on CAR T-cell functions (cytotoxicity, cytokine secretion, and cell proliferation) were investigated. Two different CAR T cells (CD133-specific CAR and HER2-specific CAR) were prepared, and the corresponding target cells including human glioma cell line U251 CD133-OE that overexpress CD133 and human breast cancer cell line MDA-MB-453 were used for functional assay. We found that lenalidomide promoted the killing of U251 CD133-OE by CD133-CAR T cells, the cytokine secretion, and the proliferation of CD133-CAR T cells. Lenalidomide also enhanced the cytotoxicity against MDA-MB-453 and the cytokine secretion of HER2-CAR T cells but did not affect their proliferation significantly. Furthermore, lenalidomide may regulate the function of CAR T cells by inducing the degradation of transcription factors Ikaros and Aiolos.
Introduction
Chimeric antigen receptor (CAR) T-cell immunotherapy is one of the hottest areas of tumor immunotherapy. Its working principle is to combine the tumor antigen recognition ability of CAR with the cytotoxicity of T cells. And its process mainly contains obtaining CAR T cells through gene engineering, activating and expanding them in vitro, and then injecting them back into cancer patients to kill tumor cells specifically. Classical CAR is composed of tumor antigen-binding, hinge, transmembrane, and intracellular signal regions. The extracellular antigen-binding region is usually a single-chain variable fragment derived from the monoclonal antibody, which specifically binds tumor antigens in a major histocompatibility complex (MHC)-independent manner. The intracellular signal region consists of signal domains that trigger T-cell activation. The first generation of CAR has only one CD3Ζ domain, the second generation contains the CD3Ζ domain and a co-stimulatory domain (CD28 or 4-1BB), and the third generation contains CD3Ζ domain and two co-stimulatory domains (CD28 + OX40 or CD28 + 4-1BB). The CAR gene can be introduced into cells through retroviral or transposon systems. Its unique structure confers T-cell tumor-specific cytotoxicity and antitumor activity in an MHC-independent manner 1 .
At present, CAR T-cell therapy has achieved outstanding efficacy in the treatment of hematological malignancies. However, for patients with solid tumors, it only induced very limited response. One of the critical factors is that CAR T cells become dysfunctional or exhausted in solid tumors 2 . Therefore, new strategies to improve CAR T-cell function against solid tumors are needed. Lenalidomide (CC-5013, Revlimid; Celgene Corporation, Warren, NJ, USA) is an oral immunomodulator that has been approved by the US Food and Drug Administration (FDA) for treatment of myelodysplastic syndromes and multiple myeloma (MM) 3 . And lenalidomide may also play a role in regulating CAR T-cell activity against hematological malignancies 4 –6 . However, the effect of lenalidomide on CAR T-cell function against solid tumor cells has not been extensively studied.
In this study, two third-generation CAR (including two co-stimulatory molecules CD28 and 4-1BB) T cells, CD133-specific CAR T cells and HER2-specific CAR T cells, were prepared as effector cells. At the same time, two tumor cells, human glioma cell line overexpressing CD133 (U251 CD133-OE) and HER2-positive human breast cancer cell line (MDA-MB-453), were chosen as target cells corresponding to the two CAR T cells. Lenalidomide was used as an immunomodulator to investigate its effects on the functions of CD133-CAR T cells and HER2-CAR T cells. We demonstrated here that the combination of lenalidomide and CAR T cells had stronger antitumor effects than CAR T cells alone.
Materials and Methods
Reagents
Lenalidomide was purchased from MCE (HY-A0003, Monmouth Junction, NJ, USA), dissolved in dimethyl sulfoxide (DMSO) at a concentration of 10 mM, stored at −80°C, and used within 1 yr. TransAct was purchased from Miltenyi Biotec, Bergisch Gladbach, Germany (T Cell TransActTM, human: 130-111-160) and stored at 4°C.
Cells
Human glioma cell line U251, breast cancer cell line MDA-MB-453, and MDA-MB-468 were purchased from Cell Bank of Chinese Academy of Sciences (Shanghai, China). Wild-type U251 (U251 WT) was nucleofected with a transposon plasmid encoding human CD133 protein and a piggyBac transposase vector to get U251 CD133-OE. Then the firefly luciferase-expressing transposon plasmid and the piggyBac transposase vector were used to transfect U251 CD133-OE again to obtain tumor cell line U251 CD133-OE luc. Similarly, the tumor cell line U251 WT luc, MDA-MB-453 luc, and MDA-MB-468 luc were also obtained by nucleofection. Peripheral blood mononuclear cells (PBMCs) were bought from ALLCELLS (PB005F, Alameda, CA, USA). Following the previously described method, PBMCs were thawed and rest for 1.5 to 2 h, then 2 × 107 cells were resuspended in 100 µl of the buffer from the Human T cell Nucleofector® kit (VPA-1002, Lonza, Basel, Switzerland), and CD133-CAR or HER2-CAR piggyBac transposon plasmid and piggyBac transposase vector were delivered into T cells by nucleofection to obtain CD133-CAR T cells or HER2-CAR T cells 7 .
Cell Culture
AIM V™ (A3021002, ThermoFisher Scientific, Waltham, MA, USA) + 10% fetal bovine serum (FBS; 10099141C, ThermoFisher Scientific) and RPMI 1640 (10-040-CVR, CORNING, Corning, NY, USA) + 10% FBS + 1% P/S (penicillin-streptomycin, 15140122, ThermoFisher Scientific) were used to expand and culture CD133-CAR T cells, HER2-CAR T cells, and NT cells. The medium used for the culture of U251 CD133-OE luc and MDA-MB-453 luc cells was DMEM (10-017-CVR, CORNING) + 10% FBS + 1% P/S. The medium used for the coculture of CAR T cells and tumor cells was RPMI 1640 + 10% FBS + 1% P/S.
For CAR T cells preparation, nucleofected T cells were activated with Dynabeads™ Human T-Expander CD3/CD28 (11141D, ThermoFisher Scientific) at a ratio of 3 beads per cell on day 0, and 300 IU/ml interleukin 2 (IL-2) was added to the medium. On the 14th day, allogeneic PBMCs from five donors were irradiated at 40 Gy using an x-ray irradiator (RS2000PRO, Rad Source Technologies, Buford, GA, USA) and added to the culture of CAR T cells at a ratio of 10:1. For the second round of expansion, 300 IU/ml of IL-2 was added to the cell culture medium, and 50 ng/ml of anti-CD3 antibody (130-093-387, Miltenyi Biotec) was added as an activator. Two days after each stimulation, CAR T cells were washed and further cultured in CAR T-cell culture medium (AIM V™ + 10% FBS) with 300 IU/ml IL-2. A full or half-volume of the medium refresh was performed every 2 to 3 d and fresh IL-2 was supplemented. Seven days after the first stimulation, beads were removed, and T cells expressing CD133-CAR or HER2-CAR were selected with 0.5 μg/ml of puromycin. On day 24 (10 d after the second stimulation), transfected T cells were analyzed by flow cytometry. Nontransfected (NT) cells were obtained in the same way, but nucleofection and drug selection were avoided 8 . For the culture of tumor cells, medium exchange was performed every 2 to 3 d. Into the medium of U251 CD133-OE luc, 1 mg/ml G418 and 1 μg/ml puromycin were added, and 1 mg/ml G418 was added to the U251 WT luc, MDA-MB-453 luc, or MDA-MB-468 luc culture.
Cytotoxicity Assay
CD133-CAR T cells (or HER2-CAR T cells) were cocultured with U251 CD133-OE luc (or MDA-MB-453 luc) tumor cells in a 96-well plate according to different effectors: target ratios (104 tumor cells per well) in 200 µl medium. Lenalidomide (final concentrations of 10, 1, and 0.1 μM, respectively) and the same dilution of solvent DMSO (103×, 104×, and 105×) were added as different groups, and tumor cells were cultured separately as controls. After the appropriate time of coculture, the substrate
Cytokine Secretion
CD133-CAR T cells (or HER2-CAR T cells) were cocultured with U251 CD133-OE (or MDA-MB-453) tumor cells at an effector-to-target ratio of 2:1, and the number of tumor cells was 105/well in a 96-well plate with a volume of 200 μl. Lenalidomide with a concentration of 10 μM, DMSO (103×) and T-cell culture medium at the same dilution were added. After 24 h, the supernatant in the culture was taken, and the cytokines IL-2, tumor necrosis factor alpha (TNFα), interferon gamma (IFNγ), and granulocyte-macrophage colony-stimulating factor (GM-CSF) were detected using AlphaLISA detection kits (IL-2, AL221C; TNFα, AL208C; IFNγ, AL217C; and GM-CSF, AL216C) from PerkinElmer, Waltham, MA, USA. Then the lenalidomide group and the DMSO group with the same dilution factor were compared to determine the effect of lenalidomide on cytokine secretion.
Cell Proliferation
U251 CD133-OE and MDA-MB-453 tumor cells were irradiated with 70 Gy. CD133-CAR T cells and HER2-CAR T cells were labeled with 0.5 μM CFSE (carboxyfluorescein diacetate succinimidyl ester, 65-0850-84, ThermoFisher Scientific) according to the instruction from the manufacturer. The two CAR T cells were cocultured with the corresponding irradiated target cells in 1 ml per well in a 24-well plate according to the effector-to-target ratio of 2:1. The CAR T-cell concentration was 1 × 106/ml. TransAct was added to the medium of NT cells at 1% to activate T cells, and the NT-cell concentration was 1 × 106/ml. Finally, different concentrations of lenalidomide (final concentrations of 10, 1, and 0.1 μM in the medium) and DMSO (103×, 104×, 105×) with same dilutions were added to each group. After 4 d, the CFSE signal in T cells was detected by flow cytometry. The lower CFSE signal indicates the faster proliferation. The difference between the lenalidomide group and the DMSO control group was analyzed. And then the effects of lenalidomide on the proliferation of two CAR T cells and NT cells were determined.
Western Blot
In 24-well plates, 2 × 106 CD133-CAR T, HER2-CAR T, or NT cells were cultured, 2 ml per well, and 1 μM, 10 μM lenalidomide or DMSO was added to the medium. After 48 h, the cells were collected, washed once with PBS, and lysed by ultrasound (5 s × 3 times). The proteins on the gel were transferred onto a polyvinylidene difluoride (PVDF) membrane using the semi-dry transfer method (15 V, 30 min), and blocked with 5% bovine serum albumin for 2 h. The PVDF membrane was incubated with anti-Aiolos (NBP2-24495SS, Novus, Littleton, CO, USA), anti-Ikaros (sc-398265, Santa Cruz Biotechnology, Santa Cruz, CA, USA), or anti-GAPDH (sc-32233, Santa Cruz Biotechnology) antibody at 4°C overnight, followed by peroxidase-conjugated AffiniPure Donkey Anti-Rabbit immunoglobulin G (IgG) (H + L) (711-035-152, Jackson ImmunoResearch Laboratories, West Grove, PA, USA) or peroxidase-conjugated AffiniPure Donkey Anti-Mouse IgG (H + L) (715-035-150, Jackson ImmunoResearch Laboratories) for 1 h, and finally imaged in the Biorad Imager (Bio-Rad, Hercules, CA, USA).
Statistical Analysis
Excel was used to calculate the killing efficiency according to the formula: killing (%) = 100 × (1-fluorescence value of experimental group/fluorescence value of tumor cell alone group). FlowJo V-10 (BD Biosciences, Bedford, MA, USA) was used to determine the average number of cell division (Div. Index) in the cell proliferation experiment. Relative proliferation of T cells was calculated by following the formula: proliferation (%) = 100 × (Div. Index of experimental group/Div. Index of R-10 control group). Student’s t-test and one-way analysis of variance in Prism (GraphPad Software, San Diego, CA, USA) were used to analyze the significant difference in killing efficiency, cytokine secretion, and relative proliferation. The data were expressed as mean ± standard deviation. All experiments were repeated at least three times. In this study, P <0.05 was considered statistically significant (*, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001).
Results
Lenalidomide Enhances the Functions of CD133-CAR T Cells Efficiently
To investigate the effect of lenalidomide on the antitumor function of CD133-CAR T cells, the cytotoxicity of CD133-CAR T cells against tumor cells was first analyzed. CD133-CAR T cells were cocultured with glioma cell line U251 overexpressing firefly luciferase and CD133 (U251 CD133-OE luc) at different effector-to-target ratios. In the coculture, different concentrations (0.1, 1, and 10 μM) of lenalidomide were added, and the solvent of lenalidomide, DMSO, was used as the control. Three days later, the signal of bioluminescence produced by firefly luciferase in tumor cells was used to evaluate the viability of U251 CD133-OE luc tumor cells, and the killing efficiency was calculated. As shown in Fig. 1A, when the lenalidomide concentration was 10 μM, the killing increased as the effector-to-target ratio increased, and the killing efficiency was significantly improved in the lenalidomide group as compared with the control group, in which the same dilution factor of DMSO was added. When the lenalidomide concentration was 1 μM, the results were similar to those at 10 μM, as shown in Fig. 1B. When the lenalidomide concentration was 0.1 μM, the lenalidomide group was significantly different from the control group only when the effector-to-target ratio was the highest (2:1), and no effect of lenalidomide was observed in the case of the low effector-to-target ratio, as shown in Fig. 1C. These results indicated that lenalidomide with a high enough concentration could significantly promote the killing of U251 CD133-OE luc tumor cells by CD133-CAR T cells. Furthermore, lenalidomide would not influence the killing specificity of CD133-CAR T cells, as almost no killing of CD133-negative U251 WT luc was observed when lenalidomide was combined with CD133-CAR T cells.
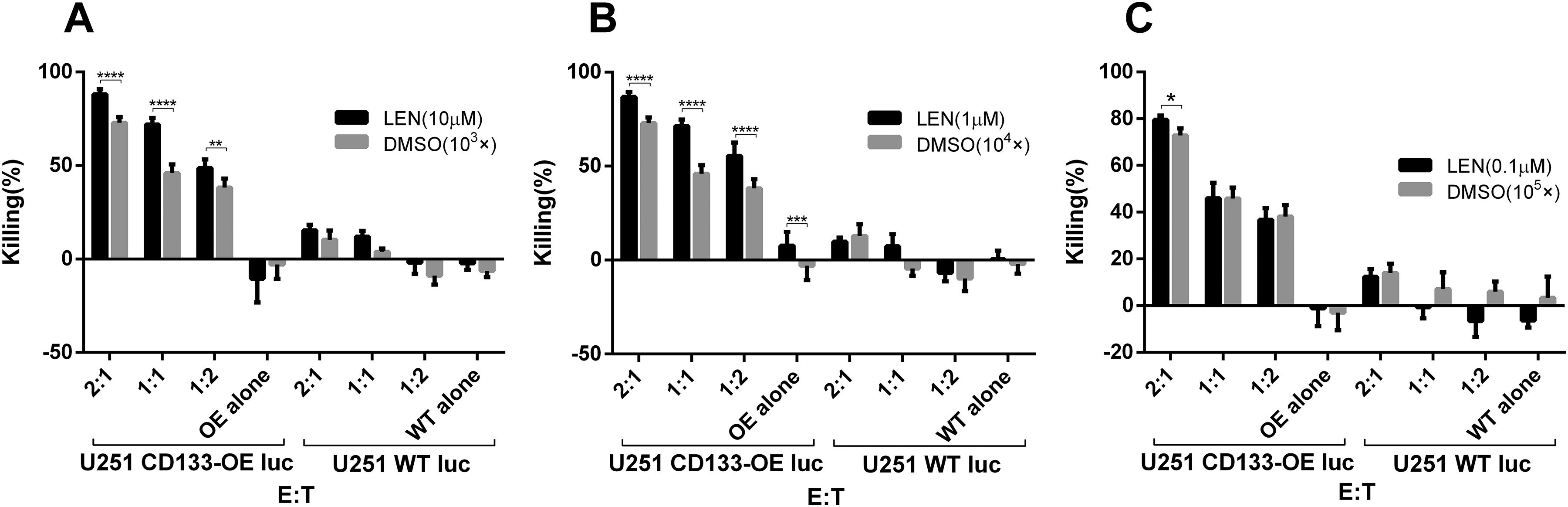
Lenalidomide promotes the cytotoxicity of CD133-CAR T cells against U251 CD133-OE luc cells. CD133-CAR T cells were cocultured with U251 CD133-OE luc cells according to different effector:target ratios. Lenalidomide (final concentrations of 0.1, 1, 10 μM, respectively) or the same dilution fold of solvent DMSO (103×, 104×, 105×) were added into the medium. After 72 h, the fluorescence signal intensity in the coculture system was detected using a microplate reader, and the killing efficiency was calculated by the formula: killing efficiency (%) = 100 * (1 − fluorescence value of experimental group/fluorescence value of tumor cell alone group). Data are shown as the mean of killing efficiency ± standard deviation in three replicates.
The effect of lenalidomide on cytokine secretion of CD133-CAR T cells was further studied. U251 CD133-OE cells were cocultured with CD133-CAR T cells, and lenalidomide was added to the medium at a final concentration of 10 μM. After 1 d, the amount of cytokine in the supernatant was measured using an AlphaLISA detection kit. As shown in Fig. 2A–D, there was no significant difference in the concentration of cytokines IL-2, TNFα, IFNγ, and GM-CSF between DMSO and T-cell medium (R-10) group. The secretion of IL-2, TNFα, and IFNγ was significantly increased in the lenalidomide (10 μM) group compared with DMSO group, as shown in Fig. 2A–C, but there was no significant difference in the secretion of GM-CSF between the two groups, as shown in Fig. 2D. This indicates that lenalidomide can promote the secretion of IL-2, TNFα, and IFNγ by CD133-CAR T cells, but does not affect the secretion of GM-CSF.

Lenalidomide enhances cytokine secretion by CD133-CAR T cell. To test the effect of lenalidomide on the cytokine secretion of CAR T cell, CD133-CAR T cells were cocultured with U251 CD133-OE tumor cells. There were three groups, and in different group lenalidomide (final concentration: 10 μM), DMSO (103×) at the same dilution or culture medium (R-10) was added. After 24 h, the secretion of the four cytokines IL-2, TNFα, IFNγ, and GM-CSF in the supernatant was detected using AlphaLISA detection kits.
Next, the effect of lenalidomide on the proliferative capacity of CD133-CAR T cells was examined. The irradiated U251 CD133-OE cells were used to stimulate CFSE-labeled CD133-CAR T cells. The ratio of T cells to tumor cells was 2:1, and the final concentrations of lenalidomide tested in the culture medium were 0.1, 1, and 10 μM, respectively. After 4 d, the CFSE signal in CD133-CAR T cells was detected by flow cytometry, and the cell proliferation was evaluated. As shown in Fig. 3A, DMSO had little effect on the proliferation of CD133-CAR T cells. However, lenalidomide (0.1, 1, and 10 μM) was found to increase the proliferation of CD133-CAR T cells compared with the DMSO group with the same dilution factor, as shown in Fig. 3B–D.

Lenalidomide promotes CD133-CAR T-cell proliferation. To study the effect of lenalidomide on the proliferation of CAR T cells, CFSE-labeled CD133-CAR T cells were cocultured with irradiated U251 CD133-OE cells. At the same time, lenalidomide (final concentrations of 0.1, 1, and 10 μM, respectively), a corresponding dilution fold of the solvent DMSO (103×, 104×, 105×), and a medium control were added. After 96 h, the CFSE signal in the coculture system was detected by flow cytometry to evaluate the proliferation of CAR T cells. (A–D) The signal value of CFSE from each group was shown, and the smaller CFSE signal intensity indicates the faster cell proliferation. (E) The relative T-cell proliferation compared to the medium control group was summarized from three independent experiments.
Lenalidomide Could Also Regulate HER2-CAR T Cells
In the above experiments, it has been made clear from the three aspects of cytotoxicity, cytokine secretion, and cell proliferation that lenalidomide can enhance the function of CD133-CAR T cells. To further explore the role of lenalidomide in other CAR T cells, HER2-CAR T cells were prepared as effector cells.
First, to investigate the effects of lenalidomide on the cytotoxicity of HER2-CAR T cells, they were cocultured with tumor cells MDA-MB-453 luc at different ratios (1:1, 1:3). Lenalidomide or DMSO with the same fold of dilution was added into the medium. The tumor signals were detected after 1 d. As shown in Fig. 4A–C, when the concentration of lenalidomide or DMSO is constant, the lysis efficiency of CAR T cells increased with the increase of the effector:target ratio. And in the case of the same effector:target ratio, the lysis efficiency of the lenalidomide group was significantly higher than the DMSO group with the same dilution factor (Fig. 4A and B), indicating that lenalidomide enhanced the killing of HER2-CAR T cells against tumor cells MDA-MB-453 luc. In addition, when HER2-negative MDA-MB-468 luc cells were used as targets, lenalidomide did not affect the function of HER2-CAR T cells.
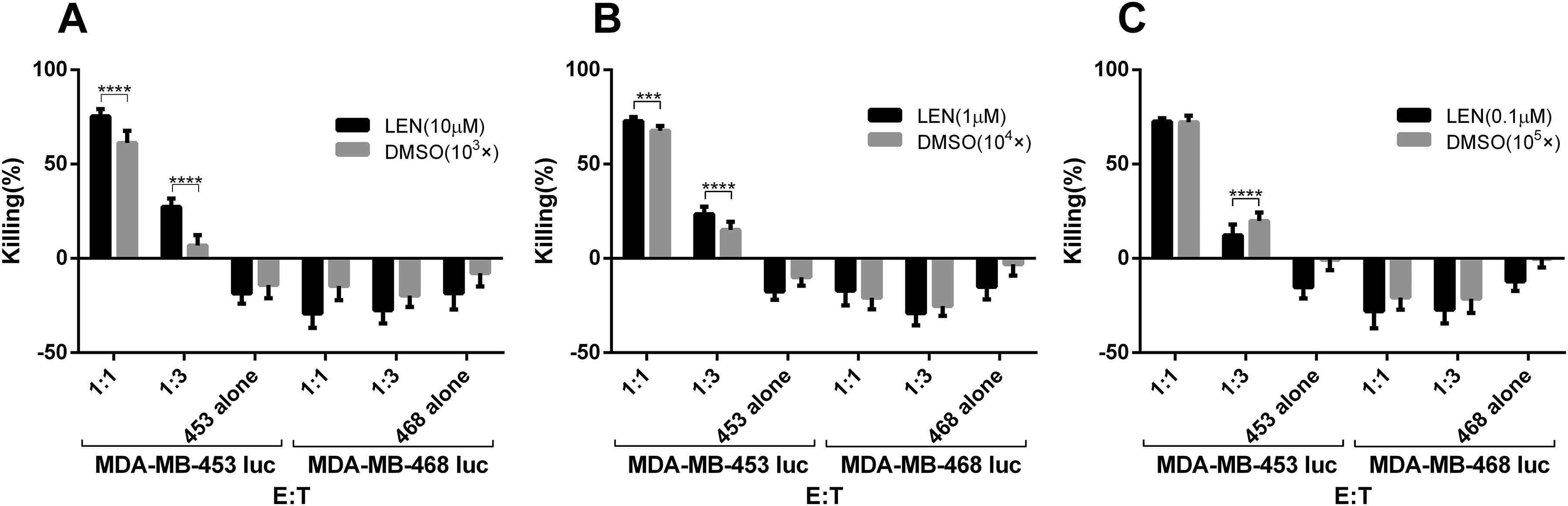
Lenalidomide increases the cytotoxicity of HER2-CAR T cells against MDA-MB-453 luc tumor cells. HER2-CAR T cells were cocultured with MDA-MB-453 luc at different E:T. Lenalidomide was added with the final concentrations of 0.1, 1, and 10 μM, respectively. After 24 h, the fluorescence intensity in the coculture system was detected using a microplate reader, and then the killing efficiency was calculated.
To know the effect of lenalidomide on the cytokine secretion of HER2-CAR T cells, the coculture was established at the effector:target ratio of 2:1 for 1 d. And 10 μM lenalidomide was tested. AlphaLISA kits were used to detect the cytokines secreted in the supernatant. The results showed that lenalidomide (10 μM) not only significantly promoted the secretion of cytokines IL-2, TNFα, and IFNγ by HER2-CAR T cells (Fig. 5A–C), but also the secretion of GM-CSF (Fig. 5D), which was not observed in the CD133-CAR T cell. Therefore, lenalidomide can promote the secretion of IL-2, TNFα, IFNγ, and GM-CSF by HER2-CAR T cells.

Lenalidomide can also enhance cytokine secretion of HER2-CAR T cells. MDA-MB-453 tumor cells and HER2-CAR T cells were cocultured according to the E:T of 2:1. And then 10 μM lenalidomide or solvent DMSO was added. After 24 h, the amount of cytokines secreted in the supernatant was detected. (A–D) Show the secretion of IL-2, TNFα, IFNγ, and GM-CSF, respectively.
Next, to investigate the effect of lenalidomide on the proliferation of HER2-CAR T cells, CFSE-labeled HER2-CAR T cells and irradiated tumor cells MDA-MB-453 were cocultured. Different concentrations of lenalidomide or DMSO were added. After 4 d, signals of CFSE in HER2-CAR T cells were detected. As shown in Fig. S1, neither DMSO nor lenalidomide could affect the proliferation of HER2-CAR T cells.
Lenalidomide Regulating the Function of T Cells Could Be CAR Independent
The above results show that lenalidomide has different regulatory effects on two different CAR T cells: CD133-CAR T cells and HER2-CAR T cells. However, in all experiments, CAR T cells are activated in the presence of tumor cells through CAR to recognize tumor antigen, thereby inducing antitumor functions. The above experiments cannot exclude the interference of target cells. To this end, NT control T cells were prepared, and TransAct (T Cell TransActTM, human) was used as the T-cell activator. TransAct would activate NT cells through CD3 and CD28 instead of the CAR activation. Then the effects of lenalidomide on cytokine secretion and cell proliferation of NT cells were examined.
First, the activated NT cells were divided into three groups, and lenalidomide was added to the first group at a final concentration of 10 μM. The second and third groups contained DMSO and T-cell culture medium. After 24 h, the supernatant was taken, and the cytokine was detected. Compared with the T-cell culture medium group, DMSO (103×) had almost no effect on the secretion of the cytokines IL-2, TNFα, IFNγ, and GM-CSF; but lenalidomide (10 μM) could significantly promote NT cell to secrete more IL-2, TNFα, IFNγ, and GM-CSF, as shown in Fig. 6A–D. This is similar to the results observed in HER2-CAR T cells.

Lenalidomide promotes cytokine secretion of NT cells. TransAct was used to activate NT cells, and lenalidomide (final concentration of 10 μM) or an equivalent diluted DMSO (103×) was added. After 24 h, the secretion of four cytokines IL-2, TNFα, IFNγ, and GM-CSF in the supernatant was detected, and the effects of lenalidomide and DMSO on cytokine secretion were compared and analyzed.
CFSE-labeled NT cells were then used to study cell proliferation ability. And TransAct was added as an activator to NT-cell culture. After 4 d, the CFSE signal in NT cells was detected by flow cytometry. DMSO had almost no effect on the proliferation of NT cells, as shown in Fig. S2. And two concentrations of lenalidomide (0.1, 1 μM) showed inhibition of NT-cell proliferation.
Lenalidomide Did Not Influence Antigen Expression on Tumor Cells but Could Induce the Degradation of Ikaros and Aiolos in CAR T Cells
To investigate whether the enhanced functions of CAR T cells was caused by antigens or ligands on tumor cells, U251 CD133-OE or MDA-MB-453 was treated by lenalidomide. One day later, tumor antigens (CD133 or HER2) and the important immune checkpoint ligand PD-L1 on tumor cells were detected by flow cytometry. As shown in Fig. S3, the expression levels of CD133, HER2, or PD-L1 had not been changed by lenalidomide treatment compared with DMSO group.
It is reported that lenalidomide can specifically mediate the connection between CRBN–DDB1 ubiquitin ligase complex and transcription factors Ikaros (encoded by IKZF-1) and Aiolos (encoded by IKZF-3), which will be ubiquitinated and degraded later, and then promote the secretion of IL-2 from T cells 9,10 . Therefore, we were wondering whether lenalidomide regulated the expression of Ikaros and Aiolos in CAR T cells we used in this study. CD133-CAR T cells, HER2-CAR T cells, or NT cells were treated by lenalidomide or DMSO for 48 h, then were lysed by ultrasound, and the expression levels of Ikaros and Aiolos were measured by western blot. We found that lenalidomide indeed induced the significant decrease of Ikaros and Aiolos expression in unstimulated CD133-CAR T cells, HER2-CAR T cells, or NT cells (Fig. 7).

Lenalidomide induced the degradation of Ikaros and Aiolos in T cells. CD133-CAR T cells, HER2-CAR T cells, or NT cells were treated with lenalidomide at 1 and 10 μM for 48 h. The cells were collected, lysed by ultrasound, and the expressions of Aiolos, Ikaros, and GAPDH were detected by western blot.
Discussion
In this study, we found that lenalidomide could regulate the function of T cells activated in different ways, including CD133-CAR T cells and HER2-CAR T cells activated by CAR signals, and NT T cells activated in a non-CAR way (TransAct). First, lenalidomide could promote CD133-CAR T cells killing U251 CD133-OE tumor cells, secreting IL-2, TNFα, IFNγ, and cell proliferation. Second, lenalidomide also promoted the cytotoxicity, and the secretion of IL-2, TNFα, IFNγ, and GM-CSF by HER2-CAR T cells, but had almost no effect on their proliferation. At last, lenalidomide could promote the secretion of cytokines IL-2, TNFα, IFNγ, and GM-CSF by NT cells, but significantly inhibit their proliferation. The mechanism of this difference needs further study.
Although the exact antitumor mechanism is not completely clear, extensive research has found that lenalidomide has multiple effects against tumors, including direct inhibition of tumor growth and indirect antitumor effects on the tumor microenvironment. Direct toxicity of lenalidomide against tumors includes inducing cancer suppressor genes 11 , causing actin polymerization, relocation of membrane proteins, 12 and cytoskeleton regulation 13 , eventually leading to cell cycle arrest 11,14,15 and tumor cell apoptosis. No direct effect of lenalidomide on tumor cells was observed in this study.
The impact on the tumor microenvironment mainly includes the following four aspects. First, lenalidomide changes the tumor microenvironment through anti-angiogenesis 16,17 . Second, lenalidomide promotes the recovery of immune synaptic defects, which has been observed in a variety of B-cell malignancies, such as chronic lymphocytic leukemia 12,18 and follicular lymphoma 19 . Third, lenalidomide increases the cytotoxicity of natural killer cells, and then enhances antibody-dependent cell-mediated cytotoxicity 11,20,21 and suppresses the function of regulatory T cells 22 . At last, lenalidomide has strong T-cell co-stimulation properties. Lenalidomide promotes T-cell proliferation and increases the secretion of IL-2 and IFNγ by activated T cells 23,24 , which is consistent with the phenomenon observed in this study.
Related studies on molecular mechanisms have found that Cereblon (encoded by the gene CRBN) is a target for lenalidomide in the treatment of MM and can mediate the co-stimulation effect of lenalidomide on T cells. Cereblon combines with DDB1 (damaged DNA binding protein 1), CUL4A (Cullin 4A), and ROC1 (Cullins 1 regulator) to form the E3 ubiquitin ligase complex (CRL4–CRBN). This complex uses ubiquitin to label specific proteins, which are then degraded 25,26 . Kronke et al. reported that lenalidomide could promote the CRBN–DDB1 ubiquitin ligase complex to selectively bind to the transcription factors Ikaros (encoded by IKZF-1) and Aiolos (encoded by IKZF-3) and induce their ubiquitination and degradation 10 . IKZF-1 and IKZF-3 are members of the Ikaros family, which includes IKZF1-5. They play an essential regulatory role in lymphocyte proliferation and development, and can selectively enhance or inhibit the transcription level of target genes. Ikaros and Aiolos are transcriptional regulators necessary for the development of B cells and T cells, and they can inhibit the production of IL-2 27 .
DNA-binding domains in Ikaros and Aiolos can recognize the promoter region of the IL-2 gene. When the promoter site is occupied by Ikaros or Aiolos, epigenetic changes in the chromatin can be induced, which can silent the IL-2 gene expression 28 –30 . Therefore, lenalidomide promotes the binding of the E3 ubiquitin ligase complex to Ikaros and Aiolos, which makes ubiquitination and degradation of them. This subsequently reduces the levels of Ikaros and Aiolos in the promoter of the IL-2 gene, which can restore the IL-2 gene transcription and promote IL-2 secretion 9,10 . The above mechanism may contribute to the enhanced functions of CAR T cells in our study, as we found that lenalidomide also caused the reduced expression of Ikaros and Aiolos in CAR T cells.
Currently, the direct relationship between the degradation of Ikaros and Aiolos and CAR T-cell function is still not clear. However, it has been reported that IL-2 directly regulates T-cell functions in many studies. Besides the well-known function of enhancing proliferation and survival of activated T cells, IL-2 binding to IL-2 receptor can induce the unregulated expression of cytolytic effector molecules such as granzyme and perforin, which is independent of the proliferation of T cells 31 . IL-2 also increases the secretion of IFNγ by T cells 32,33 . Moreover, IL-2 was approved for treatment of renal cell carcinoma in 1992 and metastatic melanoma in 1998 by US FDA 34 , and the combination of IL-2 and tumor-infiltrating lymphocytes 35,36 or TCR-engineered T cells 37,38 also showed objective responses in patients from several clinical trials. Therefore, it is possible that lenalidomide regulating CAR T-cell activity against tumor cells in our study is partially mediated by IL-2. The combination of IL-2/lenolidomide and CAR T cells may benefit patients with solid tumors.
At present, studies on lenalidomide as a single drug or in combination with other strategies to target multiple solid tumors have made some progress. As a single drug, the antitumor effect of lenalidomide was observed in a mouse model inoculated with colon cancer cells (CT26) 39 or melanoma cell line B16-F10 16 . In the clinics, metastatic renal cell carcinoma patients have sustained stable disease (SD), reduced tumor volume, and even complete remission after taking lenalidomide 40 –42 . And certain effects have also been observed in the treatment of advanced liver cancer 43 , non-small-cell lung cancer, epithelioid hemangioendothelioma 44 , recurrent ovarian cancer, and primary peritoneal cancer 45 .
The study of combining lenalidomide with other anticancer drugs to target multiple solid tumors has found that lenalidomide can restore the sensitivity of tumors to anticancer drugs. The combination of the two in in vivo and in vitro models shows a synergistic antitumor effect. For example, when lenalidomide was combined with cisplatin to target triple-negative breast cancer cell line MDA-MB-231, lenalidomide reduced the half-maximal inhibitory concentration (IC50) value of cisplatin. Compared with single drugs, their combined application significantly inhibits angiogenesis and induces apoptosis 46 . In another study, lenalidomide and 1,25-D3 (1α, 25-dihydroxyvitamin D3, the biologically active form of vitamins D) were also targeted to MDA-MB-231 tumor cells (with vitamin D resistance). The results showed that lenalidomide restored the vitamin D-resistant cell line MDA-MB-231 to be vitamin D sensitive, increased the expression of proapoptotic protein p53, inhibited the expression of antiapoptotic protein BCL-2, and induced apoptosis of MDA-MB-231 cells 47 . From another example, lenalidomide was combined with Docetaxel to treat prostate cancer using in vitro and in vivo models. Lenalidomide could significantly reduce the IC50 of Docetaxel, decrease the invasion of three prostate cancer cells (PC3, LNCaP, and DU145 cells), and inhibit anchorage-independent growth of PC3 cells. Compared with monotherapy or control group, the overall death and apoptosis of PC3 and DU145 cells treated with lenalidomide and Docetaxel were significantly increased, the tumor growth rate was reduced, and the median survival time of PC3-bearing mice was significantly extended 48 . In a clinical study, patients with stage III/IV melanoma were treated by combining lenalidomide with Darcabazine, then complete response in two patients, partial response in four patients, and SD effect in five patients were observed 49 . Other studies have reported SD for 33 mo when lenalidomide and gemcitabine were used in the treatment of metastatic pancreatic cancer 50 . Subsequent studies have shown that lenalidomide can reduce the IC50 of gemcitabine, enhance the sensitivity of pancreatic cell lines PANC-1, MIA-PaCa-2, and BxPC-3 to gemcitabine, and improve the therapeutic effect 51 . In our study, lenalidomide was combined with CAR T cells to target solid tumors, and the immune adjuvant properties of lenalidomide for CAR T cells were explored. Our results showed that lenalidomide could promote the cytotoxicity and cytokine secretion of both CD133-CAR T cells and HER2-CAR T cells, and enhance the proliferation of CD133-CAR T cells, reflecting the synergistic effects of the combination. To our knowledge, this is the first time to show the combination of lenalidomide and CAR T cells for targeting solid tumors. Subsequent experiments will be conducted on more CAR T cells targeting different types of solid tumors to expand the range of effective effects of lenalidomide. Furthermore, these in vitro findings may be transferred to a mouse model for in vivo functional study.
In summary, our study indicates that lenalidomide can regulate the functions of CD133-CAR T cells and HER2-CAR T cells to varying degrees, providing a new basis for lenalidomide to regulate CAR T cells’ function when targeting solid tumors. Small-molecule immunomodulators combined with CAR T-cell therapy may offer new hope for the treatment of solid tumors.
Supplemental Material
Supplemental Material, Fig._S1 - Lenalidomide Enhances CAR-T Cell Activity Against Solid Tumor Cells
Supplemental Material, Fig._S1 for Lenalidomide Enhances CAR-T Cell Activity Against Solid Tumor Cells by Zhixiong Wang, Guomin Zhou, Na Risu, Jiayu Fu, Yan Zou, Jiaxing Tang, Long Li, Hui Liu, Qian Liu and Xuekai Zhu in Cell Transplantation
Supplemental Material
Supplemental Material, Fig._S2 - Lenalidomide Enhances CAR-T Cell Activity Against Solid Tumor Cells
Supplemental Material, Fig._S2 for Lenalidomide Enhances CAR-T Cell Activity Against Solid Tumor Cells by Zhixiong Wang, Guomin Zhou, Na Risu, Jiayu Fu, Yan Zou, Jiaxing Tang, Long Li, Hui Liu, Qian Liu and Xuekai Zhu in Cell Transplantation
Supplemental Material
Supplemental Material, Fig._S3 - Lenalidomide Enhances CAR-T Cell Activity Against Solid Tumor Cells
Supplemental Material, Fig._S3 for Lenalidomide Enhances CAR-T Cell Activity Against Solid Tumor Cells by Zhixiong Wang, Guomin Zhou, Na Risu, Jiayu Fu, Yan Zou, Jiaxing Tang, Long Li, Hui Liu, Qian Liu and Xuekai Zhu in Cell Transplantation
Footnotes
Acknowledgments
We would like to thank the HTS Platform in SIAIS for the technical assistance.
Ethical Approval
Ethical approval is not applicable for this article.
Statement of Human and Animal Rights
This article does not contain any studies with human or animal subjects.
Statement of Informed Consent
There are no human subjects in this article and informed consent is not applicable.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the National Key R&D Program (2019YFA0111001) of China.
Supplemental Material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.