
Abstract
Lung cancer stands among the leading causes of cancer-related death in the world. Although the molecular network implicated in lung cancer development is extensively revealed, the mortality rate is only slightly improved. MicroRNAs are small, endogenous single-stranded evolutionary conserved non-coding RNAs which involve in a wide variety of biological processes including cell growth, proliferation, metabolism, and differentiation. MicroRNAs, as novel biomarkers, have multiple functions in normal lung tissue development, and aberrant expression profiles of certain microRNAs could induce lung tumorigenesis. Similar to that of protein-coding genes, microRNA expression and function are regulated by multiple factors as well as the epigenetic network including DNA methylation and histone modification mechanisms. Furthermore, microRNAs can themselves regulate key enzymes which drive epigenetic modifications and have a pivotal effect on the cell biology. In this review, we will look into the regulatory loop linkage between microRNA expression and epigenetic modifications, and then, we will discuss the effects of epigenetics on the miRNome, as well as the role of epi-microRNAs in controlling the epigenome in human lung cancer. Better knowledge of reciprocal connection between microRNAs and epigenome will help to develop novel microRNA-orientated diagnostic, prognostic and therapeutic strategies related to human lung cancer in future.
Introduction
Lung cancer is one of the important leading causes of cancer-related death around the world. Histologically, lung cancer is divided into two main groups, including non-small-cell lung cancer (NSCLC, about 85%) and small-cell lung cancer (SCLC, about 15%). NSCLC, the most common epithelial cancer, can be further sub-classified into three more subtypes, including (1) adenocarcinoma (AD), (2) squamous cell carcinoma (SCC), and (3) large-cell carcinoma (LCC). 1 Despite years of research, the overall prognosis of lung cancer has remained unclear. Currently, routine traditional diagnostics include chest imaging and sputum cytology, which can discover only 15%–20% of lung cancer prior to spread of the disease; therefore, most of the lung cancer patients are diagnosed at a late stage, so they do not have a notable chance of survival.1,2 Since the last century, the high mortality rate of patients has not been considerably improved. The two major reasons are as follows: first, the lack of effective methods to diagnose at an early stage, and second, the ineffectiveness of therapies to the advanced stage of the disease. Hence, a wide understanding of the molecular network of lung carcinogenesis is urgently needed. 2
Genetic alterations could occur at three genomic levels: at the chromosomal level, at the nucleotide level, and at the epigenetic level. These changes could result in the regulation of key genes. So, there are different mutations and expression profiles, which are not only associated with biological processes but also altered cancer development. 2
Epigenetic regulations refer to a series of biological processes which control gene expression, including DNA methylation and histone modification which are closely associated. 3 Epigenetic patterns connect genetic information to visible phenotypes. 3 These mechanisms not only regulate the protein-coding genes but also affect protein-non-coding genes. Therefore, frequently aberrant methylation of CpG islands on the promoters of oncogenes and/or tumor-suppressor genes may lead to lung cancer development. 4
Non-protein-coding RNAs (ncRNAs) have the critical role in all biological processes. MicroRNAs (miRNAs) are small non-coding RNAs which are generated from hairpin transcripts. 5 The expression of a single miRNA or a few miRNAs in the same cluster is altered in tumor cells by genetic or epigenetic mechanisms. So, the miRNAs have the ability to modulate the epigenetic patterns of multiple different keys. 6 These miRNAs are termed as epi-miRNAs which target epigenetic machinery effectors. Therefore, there is a reciprocal relationship between miRNA and epigenetic modifications in carcinogenesis. 5
The effective role of miRNAs in tumorigenesis has prompted the search and evaluation of new strategies to make use of these molecules in cancer diagnosis (Figure 1). In this review, we will summarize the recent studies about miRNAs and also the recent findings about miRNAs and epigenetic machinery.

An overview of lung cancer diagnosis traditional steps according to the National Lung Screening Trial and new molecular-based method. 7
miRNAs
RNA, as a genetic material, synthesizes all groups of proteins through the translation process. RNAs have various types of coding and non-coding molecules such as miRNAs in animals. 8 Non-coding RNAs are produced by RNA polymerase enzyme. Moreover, more than 90% of the human genome is transcribed with no protein-coding capacity. NcRNAs are involved in different biological processes, including protein synthesis, gene regulation, and inactivation of X chromosome. 9 MiRNAs are small endogenous RNAs with 19–25 nucleotides in length, which control messenger RNA (mRNA) translation into proteins. Additionally, miRNAs not only regulate the translation of approximately 30%–60% of protein-coding genes but also cooperate in the regulation of all biological processes. 10
MiRNAs: biogenesis
MiRNAs are transcribed by RNA polymerase II (RNA Pol II), generating long primary transcripts (pri-miRNA) with poly-(A)-tailed and -capped species. Then, pri-miRNAs are processed by Drosha (an RNAse type-III protein) complexes in cooperation with the Di George syndrome critical region 8 (DGCR8), giving rise to long pre-miRNAs 70–100 nt long which are transferred from the nucleus to the cytoplasm via the nuclear export protein Exportin-5 (XPO5). 11 Then, pre-miRNAs are processed by the Dicer (a ribonuclease-III protein) in collaboration with trans-activator RNA binding protein (TRBP) to generate a functional double-stranded (ds) RNA approximately 22 nt long. 12 This dsRNA includes the mature miRNA (miRNA-5p) and the complementary strand which is termed miRNA* (miRNA-3p). Then, miRNA* is normally degraded and the mature miRNA can regulate the gene expression by base-pairing to partially complementary specific sequences at the 3′-untranslated regions (3′-UTRs) of the target mRNAs. 13 There are cis-regulatory RNA elements located on 3′-UTR position which control the gene regulation. 8 Mature miRNAs recruit Argonaute protein subfamily (Ago), the RNA-induced silencing complex (RISC) catalytic components, as well as the other additional factors such as the TRBPs which form a complex known as RISC. The responsibility of this complex is the regulation of the target mRNA and inhibition of translation. 14 The Ago protein family contains four types. In mammals, Ago2 creates endonucleolytic cleavage and catalyzes degradation of target mRNAs. 8
In the recent decade, researchers have paid considerable attention toward miRNAs as a new generation of biomarkers and therapeutic targets 15 mainly due to their unique features such as petty size, supernatural gene repression role, relative stability, 5 and degree of sequence heterogeneity. 8
Up to now, about 1000 miRNAs have been recognized. Until 2008, the Sanger Center contained 8619 entries pre-miRNAs and 8273 mature miRNA in plants, primates, rodents, birds, fishes, worms, flies, and viruses (http://microrna.sanger.ac.uk/). 2 A helpful summary of miRNA databases with structural and functional annotations was collected by Nagendra Kumar Singh. 8
MiRNAs: the regulatory role
In 1993, the first miRNA was discovered, which was associated with the development of the nematode Caenor habditis elegans by regulating the lin-14 protein. Actually, it took more years to recognize miRNAs in several other organisms, such as Homo sapiens. With more studies, it was found that miRNAs are specific to tissues, greatly conserved across different species, and have important functions in all biological processes of organism. 16
In the last decade, the crucial role of miRNAs helped find their place in biology. MiRNAs regulate target genes through two mechanisms according to the level of complementarity degree between target mRNA and miRNA. In the first mechanism, mature miRNA binds to the 3′-UTR sequence of the target mRNA with absolute complementarity, which induces cleavage and degradation of the target molecule by deadenylation and decapping. 17 In this pathway, the miRNAs act as small interfering RNAs (siRNAs) which are a type of ncRNAs and produced by Dicer, usually of exogenous origin. Through the RNA interference pathway, siRNAs direct RNA cleavage. 18 Actually, most of the characterized miRNAs were unable to completely interact with their targeted sequence of mRNA, which prevented miRNA entrance into the siRNA pathway. 19 Therefore, in many cases, the mechanisms of the disruption of protein synthesis in ribosome are implied as second pathway. 14
The second mechanism is most common at the post-transcriptional level involving suppression of translation in the initiation phase because of the imperfect sequence complementarity between the miRNA and target mRNA. 20 In cytoplasm, the RISC-bound mRNAs are packed in the structures termed processing bodies (P bodies) and are degraded. 21 Unstable mRNA degradation is dedicated by adenylate uridylate (AU)-rich elements (AREs) in the 3′-UTR of mRNAs. 22 In this mechanism of mRNA turnover, miRNAs and Dicer cooperate. The complex of miRNAs, Dicer enzyme, Ago proteins, and ARE-binding proteins is needed for target mRNA decay. 23
These mechanisms could be important in several cancer pathways such as invasiveness, metastasis, tumor recurrence, and resistance to therapy. Based on these mechanisms, not only miRNAs in high abundance are able to target their available mRNA target sites and significantly affect mRNA stability in several diseases, but also less-abundant miRNAs can synergistically regulate target expression. 24
MiRNAs: targets
The most thoroughly studied importance of miRNAs is gene silencing; so, miRNAs have the ability to modulate gene transcription during the cell cycle. 14 For example, the ability of let-7 and miRNA-369-3 swings between repression and activation, depending on the cell-cycle progression. When cells are in proliferation step, these miRNAs repress the expression of their targets, but in the cell arrest level or in differentiation step, they trigger activation. 25
Taking into account all presented explanations, some miRNAs bind selectively to 5′-UTR sequence of mRNA and induce gene expression. 26 MiR-10a is a good example of miRNA-mediated activation of mRNA expression by binding to the 5′-UTR sequence of the target mRNAs. 27
Furthermore, researchers have reported four rather unusual processing mechanisms. 28 First, after Drosha processing, miR-451 enters the RISC complex by loading its precursor, and the activation phase with Dicer is skipped during the replacement of the activity of Dicer by Ago2 protein. 29 Second, Drosha-independent mechanisms include mirtrons. In initial maturation, a mirtron uses splicing machinery system to bypass Drosha cleavage, for example, miR-320. 30 Third, another mechanism is conducted by staphylococcal nuclease homology domain containing 1 (SND1), one of the components of let-7-directed RISC regulation in RAS signaling. 31 Finally, there is a significant similarity between a motif of Ago2 protein with a domain of an essential translation initiation factor (eIF4E). These two molecules compete together to bind to the domain and promote or repress translation. 32
MiRNAs: transports
According to the recent findings, miRNAs may circulate via two pathways: the first, free attachments to selected proteins (Ago) or lipids, and second, packaged within extracellular vesicles (exosomes, apoptotic bodies, and micro-vesicles). 33 Under physiological conditions, many cell types produce exosomes releasing them into the extracellular matrix. The exosomes contain proteins, mRNA, and miRNA which move from a donor cell to the recipient cell during the ‘exosomal shuttle RNA mechanism’. The aim of this mechanism is to regulate protein production in other cells. 34 So, miRNA regulatory pathways are summarized in Figure 2.

However, further explorations about the mechanisms of miRNA biogenesis will be crucial to our knowledge of the regulatory role of miRNAs, thus developing diagnostic methods and targeted therapeutic strategies.
MiRNAs and cancer
Cancer involves genetic and epigenetic alterations, which trigger an uncontrolled cell growth. Since the recent decades, scientists have focused on coding genes and their proteins and classified them into two main groups in lung cancer: (1) oncogenes and (2) tumor-suppressor genes (TSGs;31,35–63 Table 1). The ncRNAs as well as coding genes can be affected by the genetic and epigenetic alterations. Genome-wide transcriptional analysis revealed that an aberrant miRNA profiling (miRNoma) was useful to classify a variety of human tumors proposing the putative diagnostic, prognostic, and therapeutic value of these molecules. 16 So, miRNA profiling can be used to discriminate different subtypes of a particular cancer and also is applied as novel biomarkers. 6 In 2002, the first document of a link between miRNAs and cancer was found in chronic lymphocytic leukemia (CLL) with deletion of chromosome 13q14, which leads to dysregulation of miR-15 and miR-16 encoded by this locus. 64 According to human miRNA mapping, many of miRNAs were usually located at fragile sites of the chromosomal instability regions which were involved in cancer development. 50 In lung tissue, miRNA expression pattern varies from fetal to adulthood and from normal to cancerous tissue. MiRNAs as oncogenes and tumor suppressors may have key roles in lung tissue development, and ectopic expression profiles of them could induce and promote lung cancer development 2 (Figure 3).
Oncomirs and tumor-suppressor miRNAs in lung cancer.
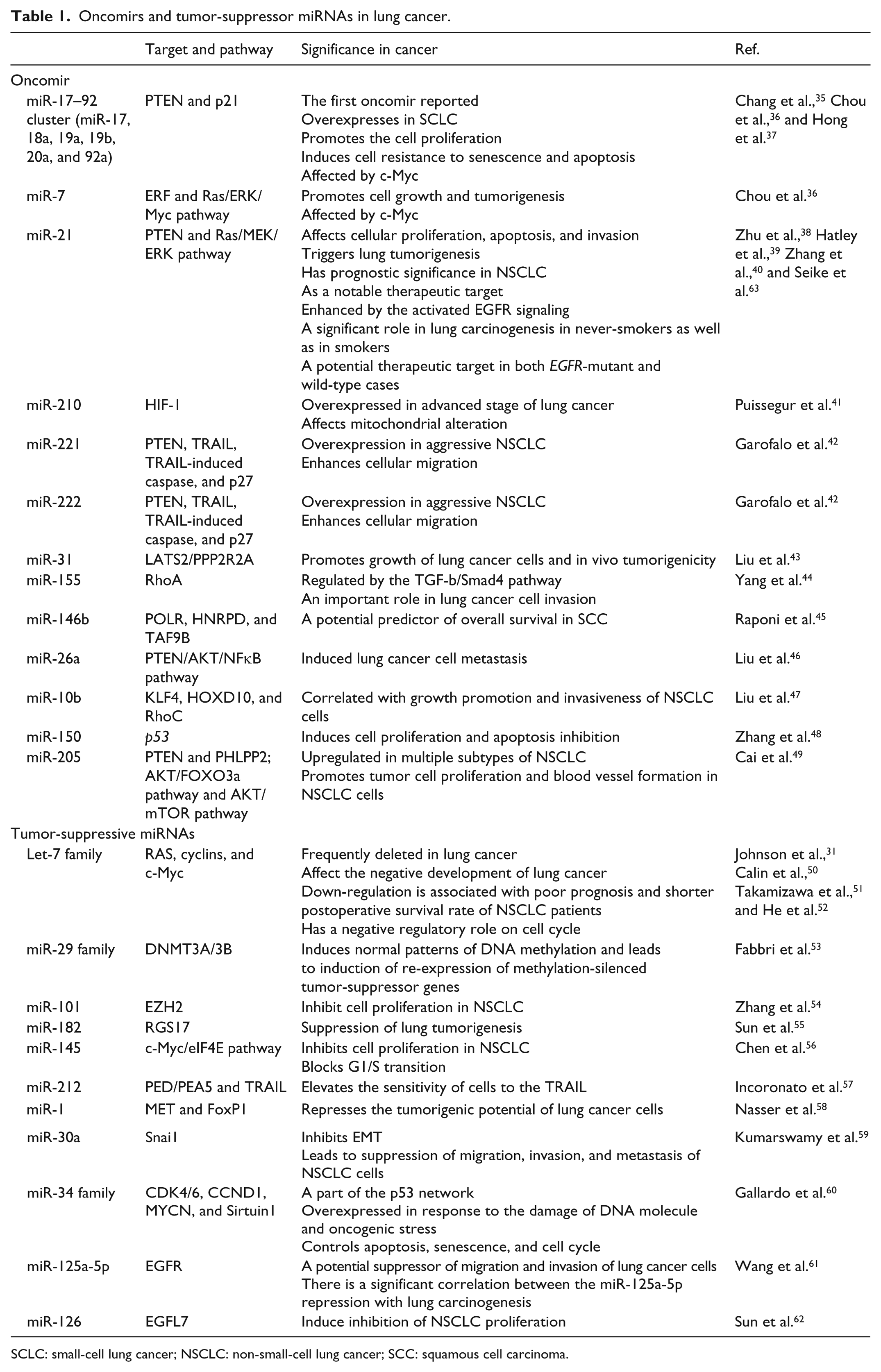
SCLC: small-cell lung cancer; NSCLC: non-small-cell lung cancer; SCC: squamous cell carcinoma.

MicroRNAs affecting oncogenic and tumor-suppressor pathways. 28
Nowadays, miRNAs can be extracted from tissue, serum, cells, and the body fluids. The optimum quality of miRNAs is identified by (1) low-throughput experimental technology (northern blotting, expressed sequence tag (EST), serial analysis of gene expression (SAGE), and Southern blotting) and (2) high-throughput techniques (deep sequencing, microarray, and real-time quantitative reverse transcription polymerase chain reaction (RT-qPCR)). 8
Epigenetics
Epigenetics is currently defined as the evaluation of heritable changes in downstream phenotypes. However, epigenetics includes structural modifications in chromosomal regions, which are independent of changes in DNA sequence. There are two major epigenetic events are involved: DNA methylation and histone modification, both of which are known to involve in gene regulation and carcinogenesis. 65 (Figure 4)
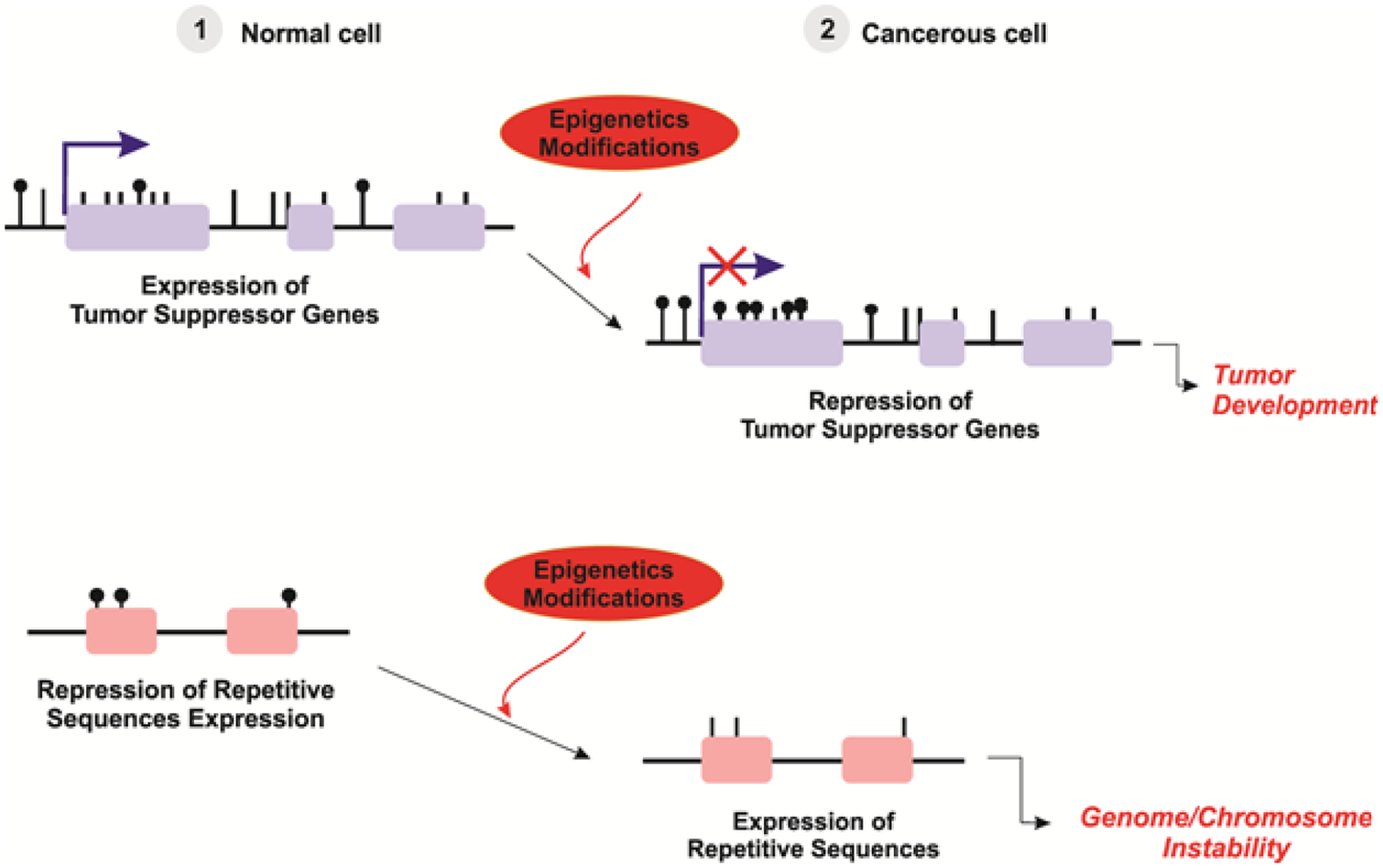
Aberrant epigenetic patterns in cancer. 14
DNA methylation
DNA methylation is characterized by the joining of a methyl group to cytosine residues and 5-methylcytosine formation. 66 Often, these modifications happen in the CpG islands, which are observed in about 60% of human gene promoters. 66 Methylation of CpG dinucleotides to a high degree leads to downregulation of gene expression. In normal cells, this is one important way to correct gene expression patterns, cell differentiation, and development. 7 Furthermore, this mechanism cooperates with the establishment of imprinting phenomena and inactivation of X-chromosome. So, high-degree methylation of repetitive sequences help to maintain chromosomal integrity. 67
There are three enzymes in human DNA methylation: DNMT1, which acts in parental methylation patterns, and DNMT3A and DNMT3B, which act in de novo methylation. Expression patterns of these enzymes are different in certain cancerous cells, but not always associated with hypermethylation/hypomethylation status. Chédin 68 partially demonstrated that expression of DNMT enzymes may be sometimes regulated by miRNA molecules.
Furthermore, the other mechanism found during the development process is the loss of DNA methylation which is catalyzed by DNA demethylases. 69 In particular, one key mechanism is carried out by hydroxylases like ten-eleven translocation 1–three proteins (TET1-3) which convert 5-methylcytosine (5mC) to the other different intermediate in the DNA demethylation process. 70 Actually, hypermethylation is usually correlated with gene silencing, while hypomethylation induces the gene activation. 69
Histone modification
Another way that gene expression is epigenetically regulated is through alterations in histone tails which are dynamic regulators of gene expression. 71 Actually, the most common histone post-translational modifications are acetylation, methylation, ubiquitylation, phosphorylation, and sumoylation. 71 Histone modifications through two mechanisms regulate gene expression; first, histone protein acetylations are correlated with transcriptional activity, and second, histone methylations lead to condensed and inactive chromatin formation. 72 Also, the enzymes which are responsible for adding and removing these groups have been expressed differentially in many cancerous cells. For example, (1) acetyl groups by histone acetyltransferases (HATs) function were added to lysine residues and removed by histone deacetylases (HDACs) and (2) methyl groups by histone methyltransferase (HMT) activity were added to lysine and arginine residues and removed by histone demethylase (HDM) enzyme. 69
Until now, the roles of these enzymes in cancer have not been completely understood. The histone modification hypothesis postulates that the expression regulation of a specific region of DNA depends upon the combination of histone protein’s modifications.
Epigenetics and cancer
Cancer can arise from a combination of genetic and epigenetic alterations, which lead to dysregulated gene expression and function. So, altered epigenetic patterns, along with genetic abnormalities, can give us a great understanding of the tumorigenesis process. 73 Actually, the most well-known epigenetic regulations existing in cancer are aberrant methylations of CpG islands, especially in promoter regions, and deacetylation/methylation alterations of histones. 73 Compared to normal cells, cancerous cells are widely hypomethylated which can mediate the overexpression of oncogenes, while most of the tumor-suppressor genes are often silenced because of hypermethylation. 69 Indeed, epigenetic machinery controls gene expression and cooperates in different cellular/physiological process; dysregulation of this mechanism in malignancies can be a preliminary transforming event and initiates genetic instability. In the case of mutation in a single allele of a gene, the epigenetic modifications could affect the other allele of the same gene through “second hit” mechanism, consequently leading to loss of heterozygosity (LOH). Finally, LOH phenomena induce inactivation of the key tumor-suppressor genes and activation of oncogenes to promote cell overgrowth and tumor development. 16
The discovery of the cooperation of epigenetic modifications in tumor development has been of considerable importance, and it can be hallmarks of cancer. Furthermore, epigenetic markers could be applied as biomarkers for many different reasons: first, isolation of genomic DNA for evaluation of methylation profiling is easier, and second, the stability of DNA molecules is more than that of mRNA for analysis of gene expression. 69
In the last years, the overlapped network of gene expression and epigenetic modification of tumor development has been complicated by the discovery of the key role of miRNAs which are able to directly regulate target genes. Furthermore, there is a straight connection between the epigenetic mechanisms and gene regulation. 16
Epigenetic modifications and miRNA expression
In recent years, the number of miRNA studies has grown rapidly in lung cancer, having proposed the crucial role of miRNAs in lung cancer development and their capability as diagnostic, prognostic, predictive, and therapeutic markers. 2 In human cancers, CpG island’s promoter hypermethylation is one of the most common causes of the silencing of tumor-suppressor miRNAs. 74 Furthermore, cancerous cells could be affected by DNA hypomethylation. The results of epigenetic modifications in tumoral cells are genome instability, chromosomal rearrangements, and genomic DNA disruption 14 (Table 2; Figure 5).
Reciprocal relationship between miRNA expression and epigenetic modifications.

EMT: epithelial-to-mesenchymal transition.

Interaction of microRNAs and epigenetic regulators. 13
Epigenetic modifications affect miRNA expression
DNA methylation pattern of tumor cells could affect miRNA expression and can be used as novel hallmark biomarker to classify tumor type, clinical diagnosis, prognosis and response to therapy. Also, histone modifications are able to affect miRNA expression profiles associated with silencing. 90
There are some methods for the detection of the miRNA epigenetic status: (1) most of the studies used pharmacologic unmasking strategy for the identification of epigenetically altered miRNAs based on microarray approach and RT-qPCR. (2) The other set of studies used the double knockout (DKO) method for DNMT1/3b. So, DNA methylation was evaluated by (1) methylation-specific PCR (MSP), (2) bisulfite genomic sequencing PCR (BSP), (3) combined bisulfite restriction analysis (COBRA), (4) bisulfite pyrosequencing, (5) methylation-sensitive single-nucleotide primer extension (Ms-SNuPE), and (6) MassARRAY assays 91 (Figure 6).

Unmasking strategy for detection of the miRNA epigenetic regulation. 91
Recent evidences suggest that cigarette smoking, as the most potential risk factor for lung cancer, affects human bronchial epithelial cells (HBECs) and induces the epigenetic silencing of specific miRNAs and leads to the decrease in the expressions of certain miRNAs and epithelial-to-mesenchymal transition (EMT).80,84
miR-9 family
In the human genome, miR-9 is located at three distinct loci: miR-9-1/2/3. So, except miR-9-2, all of them have CpG islands; 78 In all normal tissues, miR-9 family was always unmethylated. Lujambio et al. 74 treated metastatic cancer cell lines with the DNMT inhibitor 5-aza-2′-deoxycytidine and measured the miRNA expression. They reported that the DNA methylation of miR-9-3 CpG island was associated with the metastasis of human lung cancer. Kitano et al. found that the DNA methylation of miR-9-3 was associated with an advanced T factor. They found a similar proportion of methylated miR-9-3; however, they did not observe any correlation between the methylation status of miR-9-3 and lymph metastasis. 78 Heller et al. 83 evaluated the miRNA expression profile in A549 cells, which had been treated with both the tricostatin A (TSA) and the 5-aza-2′-deoxycytidine. They found that miR-9-3 could be a target for DNA methylation in NSCLC. Wang et al. evaluated the role of miR-9-3 demethylation in NSCLCs and reported that methylation silencing of miR-9-3 may affect epigenetic mechanisms in tumor cell growth, survival, and cancer progression. So, they proposed that demethylation of miR-9-3 cooperates with the anti-cancer properties of 5-AzaC and can apply as a new therapeutic strategy in the NSCLC treatment soon. 85 Tan et al. observed that methylation levels of the miR-9 were higher in NSCLC. Furthermore, miR-9-1 and miR-9-3 were also found to be correlated with age, implying these miRNA hypermethylation could be further affected the risk in aged NSCLC patients. Additionally, their study is the first study to propose that miR-9-2 hypermethylation pattern plays an important role in lung cancer patient’s survival. 86
miR-34 family
The miR-34 family consists of three miRNAs: miR-34a, miR-34b, and miR-34c which are located on chromosomes 1 and 11. 92 In mice, miR-34a and miR-34b/c are mainly expressed through the brain and lung. 93 The miR-34 family by the downregulation of multiple targets like Bcl-2, CyclinD1/E2, CDK4/6, and Myc 94 induces cell-cycle arrest and apoptosis. Moreover, this family has been demonstrated as targets of the p53 transcription factor. 93 According to these results, miR-34 family plays a crucial role as the tumor suppressor, 95 and most importantly, miR-34b/c were always unmethylated in all normal tissues. 74 Through the epigenetic alterations in lung cancer, miR-34a and miR-34b/c are silenced by DNA methylation.60,77,79,81 Furthermore, miR-34b/c is methylated in 41% of primary NSCLC 81 and 67% of the primary SCLC patients 82 and is associated with a poorer prognosis. 79 Lujambio et al. 74 treated metastatic cancer cell line with the DNMT inhibitor 5-aza-2′-deoxycytidine and reported that the DNA methylation of miR-34b/c was correlated with the human lung cancer metastasis. Watanabe et al. 81 measured the expression level of 55 in silico selected miRNAs treated with 5-aza-2′-deoxycytidine and proposed the DNA methylation–silenced miR-34b/c in NSCLC. Tanaka et al. transected miR-34b/c to SCLC which resulted in the significant inhibition of growth and invasion compared with control transfectants. Their results demonstrate that the ectopic methylation of miR-34b/c has an important role in the pathogenesis of SCLC. Actually, in SCLC, miR-34b/c can be a useful therapeutic target. 82 Furthermore, Tan et al. 86 reported that miR-34b and miR-34c are often hypermethylated in cancer tumors compared with normal tissues.
miR-148a
It is reported that miR-148a expression is important in tumor development. Studies have demonstrated that miR-148a is usually downregulated in different types of human cancers and has some crucial functions, including suppression of tumor growth, invasion, angiogenesis, and induction of apoptosis. 88 MiR-148a was always unmethylated in all normal cells. Lujambio et al. used the DNMT inhibitor 5-aza-2′-deoxycytidine for treating metastatic lung cancer, and they evaluated the miRNA expression and reported that there was a correlation between the DNA methylation of miR-148a and human lung cancer metastasis. By using computational prediction, they found that the TGIF2 gene was one of the best targets for miR-148a. 74
miR-193a
It has been found that miR-193a modulates expression of specific oncogenic factors. Heller et al. 83 treated A549 cells with 5-aza-2′-deoxycytidine and TSA. Then, they measured miRNA expression profile and identified miR-193a to be a good target for DNA methylation in NSCLC. Wang et al. evaluated the effect of miR-193a demethylation on proliferation and apoptosis in NSCLCs and found that methylation silencing of miR-193a may affect epigenetic mechanisms in NSCLC cell growth, survival, and progression. So, demethylation of miR-193a contributes, at least, to the anti-cancer properties of 5-AzaC and can be a new therapeutic strategy in the NSCLC treatment. 85
miR-126
miR-126 is located within the intron of EGFL7 gene and silenced by the DNA methylation of its host gene in NSCLC. 81 miR-126 inhibition of EGFL7 is proposed to reduce proliferation of cells in NSCLC. 86 miR-126 acts as a tumor-suppressive miRNA and through targeting Crk gene inhibits the invasion of the NSCLC. 96 In addition, miR-126 downregulation is significantly related with a shorter survival of NSCLC patients. 97 In contrast, Donnem et al. 98 reported that high expression of miR-126 is correlated with a shorter survival period and increase in vascular endothelial growth factor A (VEGF-A) expression in NSCLC patients. Watanabe et al. analyzed the expression profiles of 55 in silico candidate miRNAs treated with 5-aza-2′-deoxycytidine. They proposed that miR-126 was silenced by DNA methylation in NSCLC. 81 Tan et al. 86 showed that miR-126 is hypermethylated in tumor tissues compared to normal tissues and would result in loss of EGFL7 inhibition causing increased cellular proliferation in lung cancer.
miR-124 family
In human genome, miR-124 is located at three distinct loci: miR-124-1/2/3 embedded within CpG islands. The methylation of miR-124 is previously reported in lung cancer. Exhibited silencing of miR-124a conducted CDK6 activation and phosphorylation of retinoblastoma gene. Lujambio et al.74,76 found that CDK6 and BCL6 are the important targets for miR-124a, and epigenetic downregulation of miR-124a in cancerous cells leads to CDK6 upregulation, which is involved in cell-cycle progression and differentiation. Kitano et al. revealed that miR-124-2 and miR-124-3 showed similar methylation profiles in NSCLC specimen that were distinct from miR-124-1. The methylation statuses of miR-124-2 and miR-124-3 were individually related to recurrence of NSCLC. 78 Tan et al. 86 conducted research on the NSCLC tumor samples and found higher amount of methylated miR-124a, especially miR-124a-2 and 124a-3, in tumors compared with normal lung tissues.
miR-152
miR-152 is located at a single locus of DNA, which is embedded within CpG islands. The first study which focused on miR-152 methylation in lung cancer was conducted by Kitano and coworkers. They showed that the CpG island methylation of miR-152 was common in NSCLC. However, no considerable correlation was observed between the methylation of miR-152 and clinic-pathological features. Therefore, miR-152 may not have a dominant role in lung cancer. 78
miR-200family
The miR-200 family consists of five members: miR-200a, -200b, -200c, -141, and -429, which are expressed in two genomic clusters: chromosome 1p36.33 and chromosome 12p12.31. 99 The regulatory region of miR-200b and -200c consists of CpG-rich sequences. So, miR-200b and miR-200c were implicated in the dedifferentiation of HBEC and also in primary lung tumors. 80 The miR-200 family, by targeting ZEB1 and ZEB2 and consequently conserving E-cadherin junctions, acts as negative regulators of the EMT and prevent tumor progression. 100 Tellez et al. 80 found that the exposure of HBECs to tobacco smoking decreased the expression of miR-200b/-200c through DNA methylation mechanisms and finally induced EMT. As a result of epigenetic modification of the promoter region, miR-200c expression is decreased, and it has been associated with a poor grade of differentiation and induced an aggressive and invasive phenotype of NSCLC. 99
Let-7a-3
Let-7a-3 has an unusual oncogenic role unlike the most other let-7 miRNAs which act as tumor suppressors. 101 Let-7a-3 gene, on chromosome 22q13.31, is correlated with CpG islands. According to the study of Brueckner et al., 75 Let-7a-3 is methylated in normal lung but hypomethylated in AD. Let-7a-3 can be affected by DNMT1 and -3B, and the high expression of let-7a-3 in human lung cancer cell lines is able to induce enhanced tumor phenotypes.
miR-127
miR-127 can affect BCL6 and modulates DNA damage-induced apoptotic responses. Epigenetic silencing of miR-127 induces the activation of BCL6. 102 Tan et al. 86 reported that the hypermethylation of miR-127 is correlated with an increased risk of death and shorter survival in NSCLC, and they suggested that the epigenetic status of miR-127 could be a biomarker for the prediction of NSCLC patients’ outcome.
miR-487b
Xi et al. examined the effects of cigarette smoking on miR-487b, a novel tumor-suppressor miRNA, in cultured respiratory epithelial and lung tumor cells of two groups: smokers and nonsmokers, and they reported that cigarette smoke induces downregulation of miR-487b and thereby upregulating its targets like SUZ12, BMI1, WNT5A, MYC, and KRAS. This mechanism promotes proliferation, invasion, tumorigenicity, and metastasis of lung cancer. 84 Subsequent studies confirmed notable downregulation of miR-487b in primary lung cancer, especially in smokers, compared to adjacent normal lung tissues. In cultured cells, repression of miR-487b via cigarette smoke coincided with epigenetic modification and recruitment of polycomb repressor proteins to the regulatory region of miR-487b. 103
miR-205
miR-205 located at the region of chromosome is amplified in lung cancer. 2 Relative quantification of miR-205 can be a promising diagnostic tool and the distinction between AD and SCC. 104 Actually, miR-205 affects the epithelial cells of lung by targeting ZEB1 and ZEB2 directly and E-cadherin indirectly. 100 Furthermore, miR-205 was implicated in the HBECs’ dedifferentiation and also in primary lung tumors. Tellez et al. 80 studied the role of EMT and epigenetic silencing of miR-205, a tumor-suppressive miRNA, which affected the primary lung tumor dedifferentiation after exposure to tobacco smoking.
miRNAs which target epigenetic machinery
Not only miRNA expression is regulated by epigenetic machinery, but also it is now clear that the epigenetic modifications are targeted by miRNAs. 92 MiRNAs have the capability to regulate the expression of important epigenetic regulators, including DNMTs and HDACs, and to create an exact controlled feedback mechanism. This type of miRNA is called “epi-miRNAs,” and their dysregulated expression has been usually related to the development of human cancers.13,16
miR-29 family
The first observed epi-miRNA in lung cancer is miR-29 family. 53 The miR-29 family, including miR-29a (on chromosome7), miR-29b, and miR-29c (on chromosome 1 and chromosome 7), is upregulated in normal cells and downregulated in human lung cancer.105,106 So, miR-29a and miR-29b through the regulation of the Src-ID1 pathway act as anti-proliferative and anti-metastatic miRNAs in lung cancer. 107 So, the miR-29 family target DNMT3A/3B directly 53 and DNMT1 by targeting Sp1 indirectly. 108 The induction of miR-29s expression in lung cancer cell lines has some effects: (1) restoration of the normal pattern of epigenetic modifications, (2) reactivation of methylated silenced tumor suppressors, and (3) repression of tumor development process. 53
miR-148a
miR-148a is a dysregulated miRNA in lung cancer. 88 Duursma et al. 87 showed that human miR-148 inhibits expression of DNMT3b gene through a region in its coding sequence. Chen et al. studied about the epigenetic regulation of miR-148a in NSCLC. Due to hypermethylation of the miR-148a encoding region, the expression levels of miR-148a were decreased in NSCLC, which is associated with lymph node metastasis, advanced clinical stage, and shortened overall survival in NSCLC. So, enforced expression of miR-148a in lung cancer cells resulted in a considerable decline in the expression of DNMT1 and led to reduction in the DNA methylation of E-cadherin as a tumor suppressor. MiRNA-148a can be a potential therapeutic target to suppress lung cancer metastasis. 88
miR-101
miR-101 is frequently downregulated in lung cancer. Overexpression of miR-101 importantly induces anti-tumorigenic properties. MiR-101 inactivation causes the overexpression of EZH2, which leads to hypermethylation and aggressive tumorigenesis. 109 Yan et al. demonstrated that miR-101 downregulation correlated with DNMT3a overexpression in lung cancer. Ectopic expression of miR-101 significantly targeted the luciferase activity of DNMT3a and induced downregulation of endogenous DNMT3a, which finally led to the re-expression of CDH1, a tumor suppressor, via its promoter DNA hypomethylation, suppression of cell clonability, and migration of lung cancer cell lines and tissues. 89
These results clearly show a potential interplay between the miRNA expression and the epigenetic machinery and provide new insights into the molecular mechanisms of aberrant epigenetic modifications in lung cancer.
Limitation of microRNA study in clinical application and future plans in introducing microRNA as a biomarker in clinic
As computed tomography (CT) screening programs in the post-National Lung Screening Trial era, actually there is an urgent need to discover, develop, and validate new biomarkers which can both help identify high-risk subjects and distinguish the benign mass from malignant lesions and also be applied in therapy. While new genomic, transcriptomics, and epigenomic biomarkers have been explained above, we have yet to see progression from biomarker discovery in research phase to clinical application; reviewed biomarkers should be validated in several trials to confirm the validity. So, these biomarkers need to provide information about cancer risk, diagnosis, prognosis, and response to therapy that is independent of clinical and radiographic factors that have been well established for disease. In recent decades, according to significant progress in miRNA discovery, miRNAs have not reached clinical application in human lung cancer; so, ongoing investigation in this field suggests that miRNA signatures will be of value in the settings of indeterminate lung nodules, early detection, histologic classification, and response to therapy. 33 So, better understanding of the interaction of miRNAs and epigenomic will improve our knowledge of tumorigenic processes and will help us to discover effective strategies that can be used to conquer the lung cancer in the near future.
Conclusion
According to evidences reviewed here, miRNAs can be considered as a part of multilevel regulatory machinery; however, miRNAs not only are able to target certain molecules at the post-transcriptional level, 110 like the epigenetic machinery members, but also are strictly targeted by epigenetic mechanisms. According to these findings, aberrant balance among the members of this complicated network leads to lung cancer.
The discovery of miRNAs will probably change the landscape of lung cancer genetics. But more studies are required to understand the mechanisms by which miRNAs cooperate with cancer origin and development which requires bioinformatics methods. So, the wide data from several different sources will enable scientists to evaluate the affects of epigenetic modifications on the miRNA expression and their aberrant role in lung cancer.
All these recent findings open the possibility of new diagnostic and therapeutic strategies for lung cancer patients. This integrative project is ongoing and there is a need to update the database with upcoming studies.
Footnotes
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: The authors would like to thank Tuberculosis and Lung Disease Research Center, Tabriz University of Medical Sciences, Tabriz, Iran, for supporting this project (Grant No. 93/5), which was a part of PhD thesis (No. 93/4–10/3).