
Abstract
This study aimed to investigate the inactivation of the parkin gene by promoter methylation and its relationship with genome instability in nasopharyngeal carcinoma. Parkin was considered as a tumor suppressor gene in various types of cancers. However, its role in nasopharyngeal carcinoma is unexplored. Genomic instabilities were detected in nasopharyngeal carcinoma tissues by the random amplified polymorphic DNA. The methylation-specific polymerase chain reaction, semi-quantitative reverse transcription polymerase chain reaction, and immunohistochemical analysis were used to detect methylation and mRNA and protein expression of parkin in 54 cases of nasopharyngeal carcinoma tissues and 16 cases of normal nasopharyngeal epithelia tissues, and in 5 nasopharyngeal carcinoma cell lines (CNE1, CNE2, TWO3, C666, and HONE1) and 1 normal nasopharyngeal epithelia cell line (NP69). mRNA expression of parkin in CNE1 and CNE2 was analyzed before and after methyltransferase inhibitor 5-aza-2-deoxycytidine treatment. The relationship between promoter methylation and mRNA expression, demethylation and mRNA expression, and mRNA and protein expression of the gene and clinical factors and genomic instabilities were analyzed. The mRNA and protein expression levels were significantly reduced in 54 cases of human nasopharyngeal carcinoma compared with 16 cases of normal nasopharyngeal epithelia. Parkin-methylated cases showed significantly lower mRNA and protein expression levels compared with unmethylated cases. After 5-aza-2-deoxycytidine treatment, parkin mRNA expression was restored in CNE1 and CNE2; 92.59% (50/54) of nasopharyngeal carcinoma demonstrated genomic instability. Parkin is frequently inactivated by promoter methylation, and its mRNA and protein expression correlate with lymph node metastasis and genomic instability. Parkin deficiency probably promotes tumorigenesis in nasopharyngeal carcinoma.
Introduction
Nasopharyngeal carcinoma (NPC) is one of the most prevalent malignant tumors in South China, where its morbidity and mortality are ranked first globally. Recent studies have indicated that the etiology of NPC is multifactorial including genetic susceptibility, latent Epstein–Barr virus (EBV) infection, and exposure to chemical carcinogens. 1 During the development of the disease, EBV infection and multiple somatic genetic and epigenetic changes synergistically disrupt normal cell function, thus contributing to NPC pathogenesis. 1
The epigenetic changes in tumor suppressor genes (TSGs), especially DNA methylation, play an important role in the development of NPC. Many more putative TSGs involved in many functional processes including cell cycle regulation, DNA repair, apoptosis, tumor invasion, and metastasis were found to be silenced by epigenetic abnormalities in NPC.1–3 However, epigenetic alterations of the parkin gene in NPC have not been studied.
The parkin gene is located on chromosome 6q25-27. It contains 12 exons, spanning over 1 Mb and encodes a 465-amino acid protein with a molecular mass of approximately 52,000 Da. 4 The function of parkin protein with E3 ubiquitin protein ligase activity is to mark target proteins prior to proteasomal degradation in the ubiquitin–proteasome system. Functional loss of parkin protein leading to the accumulation of toxic substrates is thought to represent one of the pathogenic mechanisms of autosomal recessive juvenile parkinsonism. 4
However, parkin gene deletion and inactivation has been identified frequently in human malignant tumors such as colorectal cancer, lung cancer, leukemia, ovarian cancer, cervical carcinoma, pancreatic cancer, glioma, and so forth.5–12 Furthermore, parkin deletion in Apc mutant mice accelerated intestinal adenoma development and increased polyp multiplicity. 5 Parkin deficiency sensitizes mice to γ-irradiation-induced tumorigenesis. 13 Knockdown of parkin expression promotes the proliferation and tumorigenic properties of pancreatic cancer cells both in vitro and in mice. 11 Restoration of PARK2 significantly inhibited glioma cell growth both in vitro and in vivo. 12 Notably, parkin has been considered a new candidate tumor suppressor in various types of cancer.
In addition, the role of parkin in cancer is not limited to promote the ubiquitination of these proteins in an E3 ligase activity–dependent manner. It is involved in multiple signaling pathways and cellular processes.14,15 Parkin acts as a master regulator of the stability of G1/S cyclins by regulating cyclin–cyclin-dependent kinase complexes.16–18 Parkin expression induces the cell division cycle 2 (CDC2) protein phosphorylation, which inhibits CDC2 activity and causes G2/M cell cycle arrest in tumor necrosis factor (TNF)-α–treated HeLa cells. 19 Parkin serves as a tumor suppressor in human cervical cancer cells resistant to TNF-α–induced cell death by modulating survivin expression and caspase activity. 20 Parkin deficiency increases the proportion of pancreatic cancer cells with spindle multipolarity and multinucleation. 11 A C-terminal truncation of parkin results in a spindle assembly checkpoint defect in the S- to M-phase. 21 Parkin may inhibit the growth of human cervical cancer cells by decreasing the 40S ribosomal protein SA (RPSA) expression and inducing phosphorylation of cytokeratin 8/18. 22 Parkin, as a novel component in the p53 tumor suppression pathway, contributes to the functions of p53 in regulating energy metabolism, especially the Warburg effect, and antioxidant defense. 13 Parkin–Cdc20/Cdh1 complex deficiency results in mitotic defects and genomic instability. 23
Genomic instability, resulting in extensive DNA and chromosomal variations, is believed to be a driving force in carcinogenesis. 24 Genomic instability occurred frequently in most of the cancers.25–30 Random amplified polymorphic DNA (RAPD) polymerase chain reaction (PCR) was a good tool for detecting genomic instabilities. However, it remained unknown whether parkin resulting in genomic instability was involved in the development of NPC.
In a previous study, the analysis of an expression microarray with RNA from two NPC cell lines, before and after treatment with the methyltransferase inhibitor 5-aza-2-deoxycytidine, revealed that the mRNA levels of parkin in both cell lines were significantly upregulated after demethylation (unpublished data). Thus, parkin might be a target gene with its expression suppressed by promoter hypermethylation in NPC.
This study detected the methylation state and the mRNA and protein expression levels in NPC cell lines and NPC primary tumor biopsies. It also detected genomic instabilities in NPC tissues. It further analyzed the correlation between mRNA and protein expression of the parkin gene with methylation and genomic instabilities. The aforementioned evidence supports the hypothesis that inactivation of parkin by promoter methylation correlates with genomic instability in NPC.
Materials and methods
NPC cell lines, primary tumor biopsies, normal nasopharyngeal epithelium, and peripheral blood of patients with NPC
Five NPC cell lines (CNE1, CNE2, TW03, C666, and HONE1) and one normal nasopharyngeal epithelia (NNE) cell line (NP69) obtained from Guangxi Medical University (Nanning, China) were maintained at 37°C in the appropriate medium. 31 A total of 54 NPC primary tumor and peripheral blood biopsies were collected from the Department of Otolaryngology Head and Neck Surgery, Hangzhou First People’s Hospital (Hangzhou, China); 16 NNE tissues were obtained by tonsillectomy as normal controls. Informed consent was obtained from the donors. Diagnoses were established by experienced pathologists according to the World Health Organization classification. Biopsy samples were stored in liquid nitrogen before DNA or RNA extraction or paraffin sectioning. The study was approved by the local ethics committee of Hangzhou First People’s Hospital, China (approval ID: 201202501).
Semi-quantitative reverse transcription PCR
Preparation of total RNA, first-strand synthesis of cDNA, and reverse transcription PCR (RT-PCR) were performed as previously described. 32 All primer sequences, annealing temperatures, cycling conditions, and expected PCR product sizes are listed in Table 1. β-actin was amplified from the same cDNA sample as an internal control. The amplified PCR products were visualized after electrophoresis through 2% agarose gels, and semi-quantitative analysis was performed using Quantity One v 4.4.0 software (Bio-Rad, USA).
Primer sequence of parkin and β-actin genes for the semi-quantitative reverse transcription PCR (RT-PCR).

Immunohistochemical staining
The immunohistochemical staining procedure was as described. 33 Briefly, human tissue sections were stained for the expression of parkin (1:100; Abcam, UK) and detected by streptavidin–biotin–horseradish peroxidase complex formation. Tumor sections that were stained by isotype-matched immunoglobulin G instead of primary antibodies were used as a negative control.
The intensity of staining of tissues was scored as 0–3 (i.e. absent, mild, moderate, and intense); percentage of positively stained cells was also scored as 0–3 (i.e. 0: 0%–5%; 1: 6%–25%; 2: 26%–50%; 3: 51%–100%) The parkin protein expression score was calculated by multiplying these two scores (i.e. as indicated by the codes: 0; +, 1–2; ++, 3–5; and +++, 6–9).
Sodium bisulfite modification of genomic DNA and methylation-specific PCR
Sodium bisulfite modification of DNA was performed as previously described. 32 Bisulfite-modified DNA was amplified using methylation-specific PCR (MSP) with primer sets that specifically detected methylated or unmethylated alleles. All primer sequences, annealing temperatures, cycling conditions, and expected PCR product sizes are listed in Table 2. PCR products were separated through 2% agarose gels.
Primer sequence of parkin genes for the methylation-specific PCR.

5-Aza-2-deoxycytidine treatment
Two NPC cell lines (i.e. CNE1 and CNE2) were seeded into six-well plates at a density of 2 × 105 cells/well. The next day, the cells were treated with the DNA methyltransferase inhibitor 5-aza-2-deoxycytidine (5-aza-dC) (Sigma, USA) at 10 µmol/L for 96 h, and Iscove’s modified Dulbecco’s medium with freshly added 5-aza-dC was replaced every 24 h. Total RNA was extracted for semi-quantitative RT-PCR analysis. DNA was extracted for MSP analysis.
The RAPD
At total of 10 arbitrary 10-mer primers were designed.29,30 These primer sequences were as follows: P1: 5′-CCGGCTACGG -3′, P2: 5′-CAGGCCCTTC-3′, P3: 5′-TACGGACACG-3′, P4: 5′-AGCTTCAGGG-3′, P5: 5′-AGGCATTCCC-3′, P6: 5′- GGTCTGAACC-3′, P7: 5′-TAGGCTCACG-3′, P8: 5′-ACGGTACACT-3′, P9: 5′-GTCCTCAACG-3′, and P10: 5′- CTTCACCCGA-3′. In a total volume of 25 µL, 50 ng of genomic DNA extracted from carcinoma tissue or corresponding peripheral blood was amplified in 10-mM Tris-HCl (pH 8.3), 1.5 mM MgCl2, 50 mM KCl, 200 µM each of dNTP, 0.4 µM of each arbitrary primer, and 1.0 unit of AmpliTaq Gold DNA polymerase (Applied Biosystems, CA, USA). In total, 40 cycles of denaturation (94°C, 30 s), annealing (36°C –40°C, 1 min), and extension (72°C, 2 min) were carried out in a DNA thermocycler (Biometra, Germany). A volume of 5 µL of the PCR products mixed with loading buffer were loaded on to 2% agarose gels and electrophoresed with 100 V for 1 h. The gels were stained with ethidium bromide, and a photograph was taken under ultraviolet light.
Loss or gain of band(s) or an increase or decrease in the intensity of the signal of a band (≥50% by image analysis using the Quantity One v 4.4.0 software (Bio-Rad)) in the tumor DNA compared with corresponding peripheral blood DNA was scored as genomic instability.
Statistical analysis
The statistical software package SPSS 20.0 (SPSS, IL, USA) was used in this study. Data were expressed as mean ± standard deviation. Parkin expression levels in primary tumors versus NNE tissues and parkin-methylated versus unmethylated tumors were analyzed using the Mann–Whitney U test. The correlations between parkin mRNA or protein expression levels and clinicopathological features were analyzed using the Student’s t-test or the rank-sum test. The correlations between genomic instability and parkin gene methylation, transcription, protein expression, and clinical factors were analyzed by Spearman’s correlation and multivariable linear regression. An alpha value of p <0.05 was considered a statistically significant difference.
Results
Inactivation of parkin in NPC cell lines and primary tumors
To evaluate the mRNA expression levels of parkin in NPC cell lines and primary NPC biopsies, a semi-quantitative RT-PCR assay was used. Parkin mRNA expression was detected in NP69 cells and all NNE tissues (Figure 1; NP69, 4, 5, and 6 as samples) and NPC cell lines (i.e. CNE1, CNE2, and C666) were silenced, while decreased expression was detected in TW03 and HONE1. In addition, parkin expression was frequently absent or downregulated in the 54 NPC biopsies (Figure 1; 1, 2, and 3 as samples). Also, to evaluate the protein expression of parkin in NNE tissues and NPC biopsies, immunohistochemical staining was performed. Parkin protein expression was detected in all NNE cells (Figure 2(a) and Table 3), while parkin protein expression was frequently absent or downregulated in the 54 NPC biopsies (Figure 2(b) and (c) and Table 3).

RT-PCR analysis of mRNA expression of parkin, (a) and (b) indicate and the mRNA expression levels of parkin in NPC cell lines (i.e. CNE1, CNE2, TWO3, C666, and HONE1) and the normal nasopharyngeal epithelial cell line (NP69), NPC biopsies (i.e. 1, 2, and 3), and normal nasopharyngeal epithelial (i.e. 4, 5, and 6) samples, respectively. The data are representative of two independent experiments. In addition, β-actin, normal colorectal tissue, and water were used as an internal control, positive controls (7), and blank control (8), respectively. (c) and (d) indicate mRNA expression levels of the parkin gene (N) were calculated by normalization with the NP69 gene as follows: N = (gray value of parkin gene fragment bands/internal reference gray value)/(gray value of NP69 gene fragment bands/internal reference gray value). (e) indicate a decreased level of parkin expression was seen in NPC cases compared with NNE cases (p < 0.01).

Immunohistochemical analysis of protein expression of parkin in normal nasopharyngeal epithelial tissues (NNE) and NPC biopsies. (a), (b), and (c) indicate NNE-positive, NPC-positive, and NPC-negative samples, respectively (400×).
Protein expression of Parkin in normal nasopharyngeal epithelial tissues (NNE) and NPC biopsies.

Hypermethylation of parkin in NPC cell lines and primary tumors
To investigate the promoter methylation status of parkin, MSP was performed. Promoter methylation of parkin was detected in 60% (3/5) of NPC cell lines (i.e. CNE1, CNE2, and C666) and 62.96% (34/54) of primary tumors (Figure 3; T1, T2, and T3 as samples). However, this was not found in any of the 16 cases of NNE tissues, NP69, TWO3, and HONE1 (Figure 3; N1, N2, and N3 as samples).

Methylation-specific PCR analysis of the Parkin promoter region in NPC cell lines (i.e. CNE1, CNE2, TWO3, C666, and HONE1) and the normal nasopharyngeal epithelial cell line (NP69), NPC biopsies (i.e. T1, T2, and T3) and normal nasopharyngeal primary epithelia (i.e. N1, N2, and N3) samples. The data are representative of two independent experiments. In vitro methylated DNA was used as a methylation-positive control and DNA from normal lymphocytes was used as an unmethylated positive control. Water was included as a blank control. (a) and (b) indicate CNE1, CNE2,C666 ,T1 and T2 as methylated samples,others as unmethylated samples.
Inactivation of parkin correlated with its promoter hypermethylation
To verify whether mRNA inactivation of parkin was related to promoter methylation in NPC, the methylation status of the parkin gene was detected by MSP. The methylated parkin promoter could be detected in three NPC cell lines (i.e. CNE1, CNE2, and C666) in which parkin mRNA expression was silenced. However, in the NPC cell line (TWO3 and HONE1) and NNE cell line (NP69), which expressed a normal level of parkin, only the unmethylated promoter was detected (Figures 1 and 3). Additionally, promoter methylation of parkin was detected in 62.96% (34/54) of primary tumors, of which 23 cases (i.e. T1; Figure 1) were parkin-silenced. Combined with previous RT-PCR results, a decreased level of parkin expression was seen in parkin-methylated NPC cases compared with unmethylated cases (p < 0.05, Mann–Whitney U-test; Figure 4).

mRNA expression level of the parkin gene in methylated NPC and unmethylated NPC tissues. (a) was similar to Figure 1(d). (b) indicate a decreased level of parkin expression was seen in methylated NPC cases compared with unmethylated NPC cases (p < 0.01).
To further demonstrate that parkin gene methylation was directly responsible for the loss of parkin transcription, the parkin-silenced NPC cell lines CNE1 and CNE2 were treated for 4 days with the demethylating agent 5-aza-dC, following which parkin mRNA expression was restored (Figure 5(a)), and the fully methylated status was reversed to the unmethylated status (Figure 5(b)).

Demethylating treatment with 5-aza-dC restored parkin expression and unmethylated status in CNE1 and CNE2 cells. Parkin mRNA expression levels were evaluated by RT-PCR. β-actin was amplified as an internal control. NNE and water were used as positive and blank controls, respectively. (a) indicate parkin mRNA expression was restored. Promoter methylation of parkin in CNE1 and CNE2 was evaluated by methylation-specific PCR before and after 5-aza-dC treatment. (b) indicate the fully methylated status was reversed to the unmethylated status.
Clinicopathological significance of parkin gene expression
Parkin mRNA and protein expression levels were found to be markedly associated with the lymph node metastasis (p < 0.01). However, these were not correlated with the age or sex of the patients, tumor size, tumor stage, or the histopathologic subtype (p > 0.05; Tables 4 and 5).
Correlation between parkin mRNA expression and clinical data of NPC patients.

Staging according to International Union Against Cancer (UICC).
Correlation between parkin protein expression and clinical data of NPC patients.
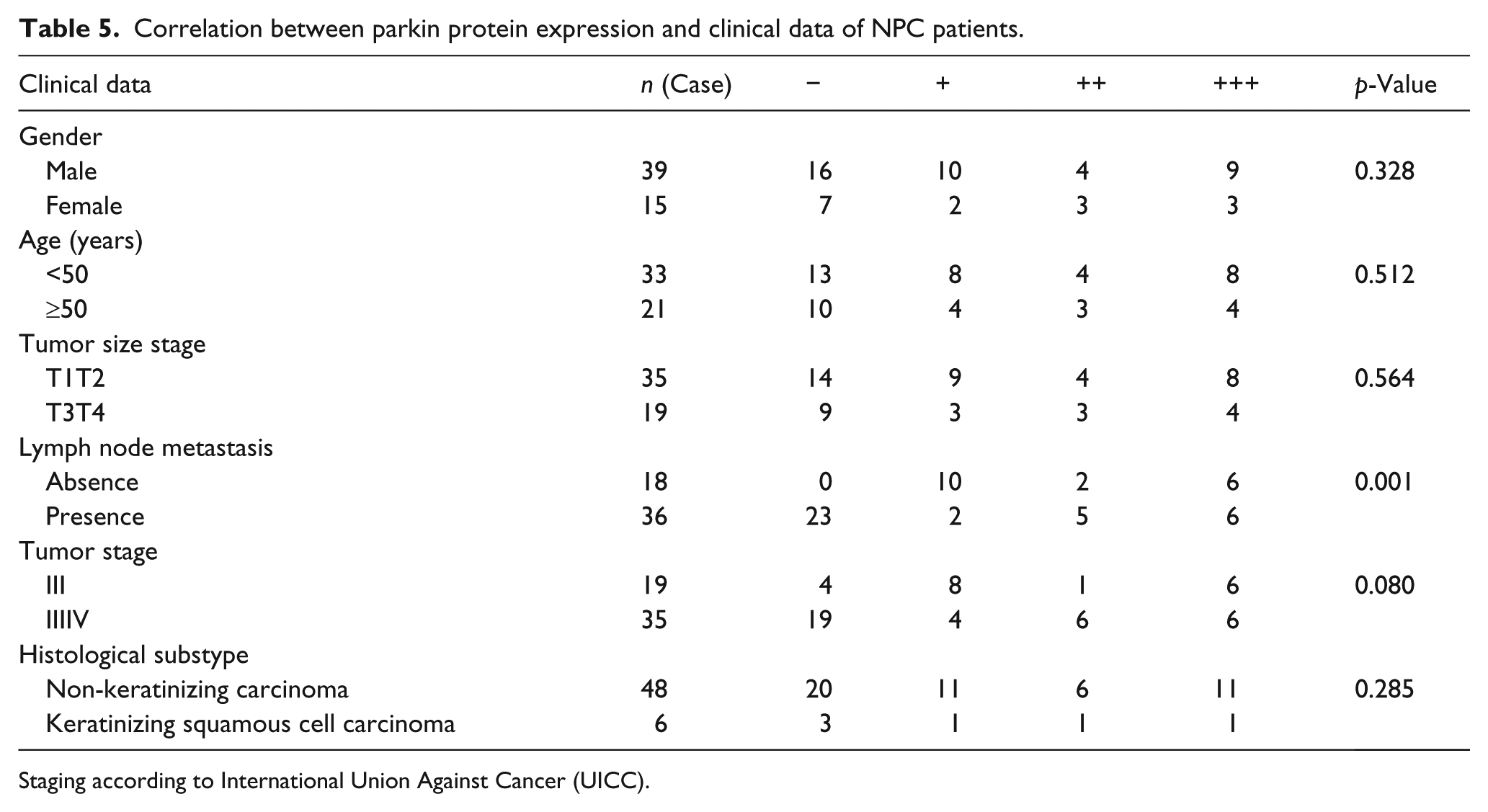
Staging according to International Union Against Cancer (UICC).
Genomic instability in NPC
Of 54 cases of NPC, 50 cases demonstrated genomic instability, which was detected by at least 1 of 10 primers. The incidence of genomic instability ranged from 0% to 100% in each case detected by 10 primers (Figure 6), and 35.19% to 83.33% in each primer (Figure 7). Loss or gain or mobility shift of a band (Figure 8(a) and (b)) and increased or decreased band intensities were observed (Figure 8(c) and (d)) in the tumor DNA compared with the corresponding peripheral blood DNA.

Incidence of genomic instability in each case of NPC detected by 10 primers.
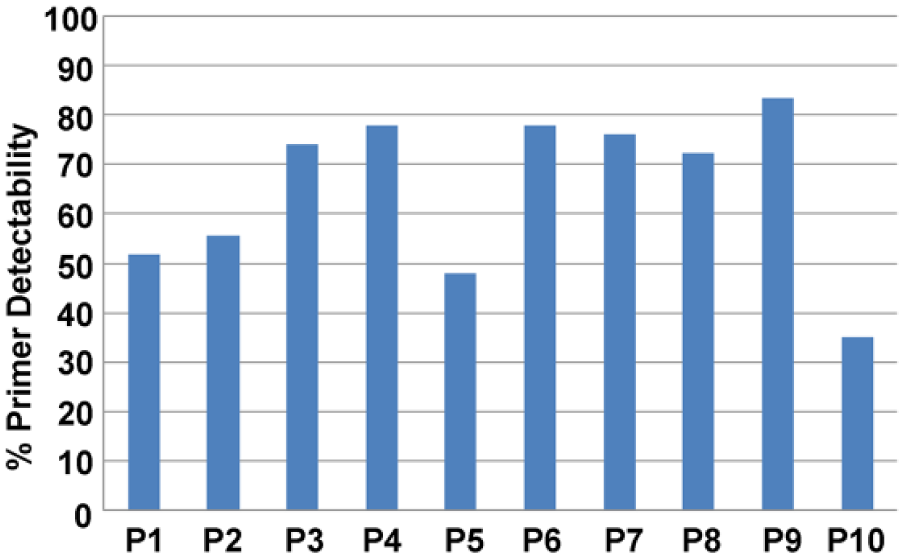
Ten primers’ detection rate of genomic instability in NPC.

RAPD PCR analysis of NPC. Tumor DNA (T1, T3, and T7) and corresponding peripheral blood DNA (N1, N3, and N7) were amplified. Cases 1, 3, and 7 with primer 10 (a), primer 8 (b), primer 3 (c), and primer 2 (d). (a, b) Loss or gain or shift of a band (arrowhead); (c, d) change in band intensity (arrowhead).
Correlation between genomic instability and parkin gene expression
Because genomic instability variables are continuous variables, linear regression analysis was performed. First, Spearman’s correlation analysis was performed between various variables (age, gender, tumor size, lymph node metastasis, tumor stage, histological subtype, methylation status, mRNA expression, and protein expression) and genomic instability. The correlation analyses of statistically significant variables were then included in the multivariate linear regression model with the significance level of 0.05.
Spearman’s correlation analysis and multivariate linear regression results suggested that genomic instability was found to have a markedly negative correlation with parkin mRNA and protein expression levels (B1 = −1.332, B2 = −1.008, p < 0.01). However, it was not correlated with parkin promoter methylation, age, or sex of the patients; tumor size; tumor stage; lymph node metastasis; or histopathologic subtype (p > 0.05).
Discussion
Parkin dysfunction has been associated with the progression of parkinsonism and human malignancies, but its role in cancer, especially NPC, is still explored.
Previous studies have considered that gene mutation and homozygous deletion are the main mechanisms of TSG transcription inactivation. 34 However, recent studies have confirmed that promoter hypermethylation plays an important role in the inactivation of cancer-related genes, and in some cases, it is the only mechanism of TSG inactivation. 35 Gene mutation and loss of heterozygosity were the predominant modes of inactivation of parkin in most cancers, such as colorectal, lung, ovarian, and other cancers.5–7,9–12,36,37 Parkin was epigenetically regulated only in human acute lymphoblastic leukemia and chronic myelogenous leukemia. 8 Thus, the role and expression of the regulatory mechanism of parkin in NPC need to be investigated.
This study confirmed that mRNA and protein expression of parkin in NPC tissues were downregulated, which indicated that parkin played a major role as a TSG in the development of NPC. The MSP results demonstrated that parkin promoter methylation was detected in 60% of the NPC cell lines and 62.96% of the primary NPC tissues, but was not detected in NP69 and NNE tissues. In other words, parkin methylation was a frequent and tumor-specific process in NPC. The MSP and semi-quantitative RT-PCR data exhibited a close correlation between mRNA expression levels and the methylation status in NPC cell lines and primary tumors. In this context, methylation inactivation appears to be a major mechanism in the loss of parkin expression. Restoration of parkin expression by the methyltransferase inhibitor 5-aza-dC further verified this possibility. Thus, parkin gene methylation has a similar role in human acute lymphoblastic leukemia, chronic myelogenous leukemia, and NPC, and yet plays different roles in most of other cancers, in which gene mutation and loss of heterozygosity were the major regulatory mechanism for the inactivation of parkin gene expression.5–7,9–12,36,37
No statistical relationship was found between parkin mRNA expression and patient age, degree of tumor grade, tumor stage, and menopausal status in ovarian carcinoma. 9 Sun et al. 11 found a significantly negative correlation between parkin protein expression and the histological grade and incidence of lymph node metastasis of pancreatic cancer. Toma et al. 38 reported that low levels of parkin mRNA were associated with high-grade clear-cell renal-cell carcinomas and lymph node metastasis, but the downregulated protein expression of parkin was not associated with the clinical outcome. However, this study demonstrated that parkin mRNA and protein expression levels in NPC tissues were significantly and inversely associated with lymph node metastasis. Low-parkin mRNA and protein expression levels might be related to lymph node invasion. This observation suggested that parkin mRNA and protein expression could be considered an indicator of lymph node metastasis in NPC. Thus, the role of parkin as a TSG in NPC is similar to that in pancreatic and clear-cell renal-cell carcinoma, but the specific mechanisms of functional gene expression of parkin in different cancers vary considerably.
Xiong et al. 6 demonstrated the PARK2 germline mutation c.823C>T (p.Arg275Trp) as a genetic susceptibility factor for lung cancer. Agirre et al. 8 found no significant differences between methylated and normal patients with regard to relapse rates, mortality rates, disease-free survival at 11 years, and overall survival at 12 years. Toma et al. 38 reported that patients with low-parkin mRNA levels showed a higher tumor-specific mortality rate and a shorter overall survival. However, most of the cases were followed up for less than 5 years, and thus the 5-year survival time of patients was not analyzed. The role of parkin in the pathogenesis, prognosis, and risk estimation of NPC may be further clarified in the future after the statistical analysis of the relevant follow-up data of the past 5 years.
Wang et al. 33 demonstrated that parkin promoted the activity of paclitaxel to trigger multinucleation and apoptosis, rendering breast cancer cells more sensitive to this drug. Another study reported that a p53 fusion protein TAT-ODD-p53 had a significant and preferential radiosensitizing effect on hypoxic breast cancer cells by inhibiting parkin-mediated mitophagy. 39 Whether parkin can improve the sensitivity of patients with NPC to radiotherapy or paclitaxel chemotherapy needs to be confirmed by further experimental studies.
The RAPD method is thought to be suitable for investigating genomic instability, which can simply and rapidly detect genetic alterations in the entire genome. 24 It is well known that genomic instability occurred frequently in most cancers.25–30 The present results also indicated the frequent occurrence of genomic instability in NPC. Furthermore, a significantly negative correlation was found between the frequency of genomic instability and the levels of parkin mRNA and protein expression; genomic instability is believed to be a driving force in carcinogenesis. 24 The present results implicated that parkin deficiency might promote tumorigenesis in NPC and hence are consistent with Lee’s findings. 23
In addition, methylation-mediated inactivation is potentially a reversible phenomenon. 40 As the parkin gene has an anticancer effect, upregulating parkin using a demethylating agent may probably prevent or reverse the malignant phenotype, and hence it may translate into a novel demethylating agent and therapeutic target in NPC.
This study revealed that parkin was frequently inactivated by its promoter methylation and its mRNA and protein expression correlated with lymph node metastasis and genomic instability in NPC, suggesting that parkin deficiency probably promoted tumorigenesis in NPC. Parkin mRNA and protein expression can be considered as an indicator of lymph node metastasis and may have a potential value in clinical applications. Whether the parkin gene can be used as a predictive indicator of the sensitivity of patients with NPC to radiotherapy and chemotherapy should be confirmed by further experiments.
Footnotes
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
This study was supported by grants from the Medical and Health Backbone Platform Foundation Project of Zhejiang Province, China (grant nos 2011KYA127 and 2012RCB038).