
Abstract
The prevalence of obesity has steadily increased over the past few decades. Previous studies suggest that obesity is an oncogenic factor and that over 20% of all cancers are obesity-related. Among such cancers, digestive system malignancies (including esophageal adenocarcinomas, colorectal cancers, and cancers of the gastric cardia, liver, and pancreas) are reported most frequently. While the 5-year survival rates of cancers of the breast and prostate are 90%, that rate is only 45% for digestive cancers. In this review, the mechanisms of obesity-associated digestive cancers are discussed, with an emphasis on obesity-related gene mutations, insulin and insulin-like growth factor signaling pathways, chronic inflammation, and altered adipokine levels. Evidence that these factors often function interdependently rather than independently in carcinogenesis is presented. Recommended interventions that may reduce the burden of obesity-associated digestive cancers, such as participation in physical activity, diet modulation, and calorie restriction, are also described.
Introduction
The prevalence of obesity has steadily increased over the past few decades. As of 2011, 500 million adults and nearly 43 million children under the age of 5 years are obese worldwide. Previous studies suggest that obesity is an important factor in oncogenesis and that over 20% of all cancers are obesity-related.1,2 In a prospective cohort study of more than 900,000 U.S. adults, overweight or obesity accounted for 14% and 20% of all cancer-related deaths in men and women, respectively. 3 A more recent cohort study proposed that an average population-wide increase of 1 kg/m2 in the body mass index (BMI) would result in an additional 3790 cancer patients in the United Kingdom every year. 4 Obesity also complicates dose determination of chemotherapeutic agents, and obese patients have higher mortality rates compared to those with other cancers.5,6
The digestive system is biochemically complex and comprises many diverse types of epithelial cells as well as different tissues with various functions that can potentially become dysregulated. The most frequently reported obesity-associated digestive malignancies include esophageal adenocarcinomas (EACs), colorectal cancers (CRCs), and cancers of the gastric cardia, liver, and pancreas.3,4,7 While the 5-year survival rates of breast and prostate cancers are 90%, those of digestive cancers are only 45%. 8
Recent studies have shown that high-fat-diet-induced obesity is a low-grade inflammatory condition, in which macrophages promote the development of insulin resistance and accompanying hyperinsulinemia.9,10 Type 2 diabetes mellitus (T2DM) exhibits similar pathophysiologic effects as obesity, including insulin resistance, hyperinsulinemia, increased bioavailable insulin-like growth factor-1 (IGF-1), and hyperglycemia.11,12 Hyperinsulinemia-induced alterations in insulin, IGF, and other signaling pathways elicit pro-carcinogenic events, 10 and there is evidence that insulin resistance and hyperinsulinemia may lead to direct activation of the insulin receptors in cancer cells, promoting tumor growth. 11 Collectively, these findings suggest that inflammation is involved in the pathogenesis of both obesity and cancer.13,14
Clearly, obesity is closely associated with tumorigenesis. This systematic review summarizes and discusses the mechanisms and effects of obesity-associated digestive cancers (Table 1 and Figure 1), as well as the interventions that might reduce the burden of these cancers.
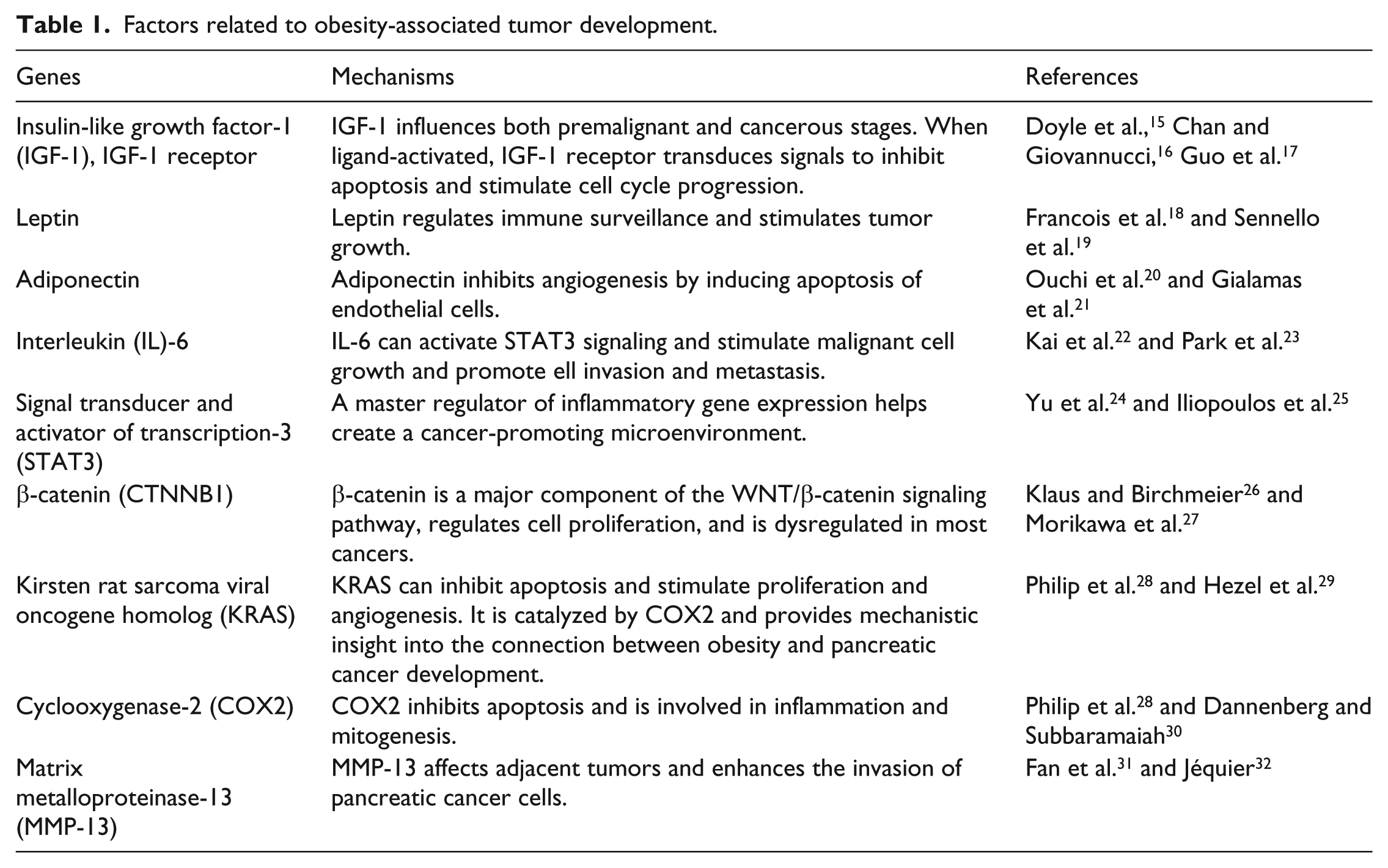
Factors related to obesity-associated tumor development.

Biological mechanism of obesity-driven tumor development. Schematic diagram represents the main mechanisms that are hypothesized to link obesity and cancer risk.
EAC
EAC usually occurs in the distal esophagus or gastroesophageal junction. 33 Its incidence has risen dramatically (as much as 600%) in the last 30 years; the disease is mostly limited to developed countries.33,34 The overall prognosis of EAC is dismal, with a 5-year survival of 15%–20% at best. 35
According to two meta-analyses, being overweight or obese increases the morbidity of EAC approximately two- to threefold or more if the BMI is high.36,37 Adenocarcinoma, rather than squamous cell carcinoma, is the most common histological type of esophageal cancer worldwide; its risk positively correlates with the BMI, most notably when the cancer is associated with obesity. 1
Obesity has been linked to gastroesophageal reflux disease (GERD) and even more strongly to Barrett’s esophagus (BE). 38 It promotes GERD by disrupting the anatomy and physiology of the gastroesophageal junction via mechanical and (more pertinently) systemic metabolic/inflammatory pathways. 39 Abdominal fat increases intragastric pressure and the frequency of transient relaxations of the lower esophageal sphincter. 40 Moreover, hiatal hernia is more prevalent in obese individuals than in normal-weight individuals and contributes to the initiation and progression of GERD. 41 Roux-en-Y gastric bypass (RYGB) surgery appears to be the preferable treatment for GERD, as alleviation of GERD symptoms after undergoing this type of surgery has consistently been demonstrated. 42
BE is an acquired condition that involves metaplastic columnar replacement of the normal stratified squamous epithelium of the distal esophagus. It is considered a premalignant disorder that occurs in response to chronic acid exposure resulting from gastroesophageal reflux. 43 BE is characterized by increased proliferative and anti-apoptotic activity, which increases the risk of genetic abnormalities and drives the development of dysplasia and malignant transformation. The epithelium in BE contains increased subsets of diploid and aneuploid cells compared to normal epithelium. 44 Upon comparing EAC patients and individuals with precancerous BE to control subjects, 19p13 in CRTC1 (that encodes the cAMP responsive element binding protein [CREB]-regulated transcription coactivator 1), 9q22 in BARX1, and 3p14 near FOXP1 were three loci found to be associated with susceptibility to these disorders. 45 Collectively, these findings suggest that GERD and BE account, at least in part, for the strong association between obesity and EAC. RYGB is not recommended as the sole means of preventing EAC 46 ; however, whether obesity directly contributes to the pathogenesis of EAC remains unknown. 47
Visceral adipose tissue shows a strong clinical association with erosive esophagitis, as its volume positively correlates with the severity of the esophagitis. 48 Obese individuals have abnormal circulating serum levels of pro-inflammatory cytokines (e.g. tumor necrosis factor-α (TNFα) and interleukin-6 (IL-6)) and adipokines (e.g. adiponectin and leptin) (Table 1), which are released from visceral fat and contribute to EAC development. 18 An elevated leptin level has been shown to be an independent risk factor for progression to cancer in a cohort of patients with BE. 49 Insulin, estrogen, and inflammatory mediators such as IL-1B, IL-6, and TNFα alter the production of gastric leptin, and variations in production can promote differential esophageal healing and metaplasia progression 18 (Figure 1). Leptin also regulates immune surveillance in gastrointestinal cancers, including EAC, by inhibiting the activity of regulatory T cells. 19
Although subcutaneous tissue contributes significantly to the production of adipokines, their primary source in obese individuals is visceral adipose tissue.50,51 Adipokine-mediated carcinogenic effects resulting from increasing visceral fat levels have been observed in several gastrointestinal cancers, including EAC. 52 In addition to releasing adipokines, visceral fat is also associated with insulin resistance, and a key role for insulin and IGF-1 in regulating the malignant progression of esophageal cancer has recently been reported 15 (Table 1 and Figure 1). Notably, obesity can complicate treatment, as can sarcopenia; obese patients with esophageal cancer have a significantly higher risk of developing limiting toxicity than do the non-sarcopenic, non-obese counterparts. 53
There is evidence associating weight loss due to lifestyle/dietary changes or surgery with a reduction in symptomatic reflux. However, whether such weight loss consequently reduces the risk of BE and EAC requires further study.47,54
Gastric cancer
Gastric cancer (GC) is the second most frequent cause of cancer-related death worldwide, affecting approximately 1 million people annually. 54 Previous studies associate obesity with an increased risk of GC, most prominently in patients with high BMIs. 55 The two main sites of gastric adenocarcinomas are the proximal and distal regions of the stomach (the cardia and non-cardia, respectively).
From 1978 to 1992, the incidence of proximal GC has increased five- to sixfold in developed countries. 56 Unlike its distal counterpart, proximal GC is generally associated with GERD and obesity. 57 Compared with tumors in the distal gastric pyloric antrum, tumors in the gastric cardia have a much poorer prognosis, with lower 5-year survival and higher operative mortality rates. 58 A Swedish study divided a sample population into quartiles based on weight and found that those in the highest quartile (BMI ≥29 kg/m2) had a 2.3-fold greater risk of proximal GC than did those in the lowest quartile (BMI ≤23 kg/m2). 59 In a U.S.-cohort study of more than 1000 individuals, a high BMI increased the risk of gastric cardia adenocarcinoma by twofold. 60 These studies clearly associated obesity with proximal GC. As stated above, obesity can promote GERD, which in turn predisposes patients to BE; all gastric cardia adenocarcinomas occur near the gastroesophageal junction, which is also where BE usually occurs.
Helicobacter pylori is a major, well-established risk factor for distal GC. 61 Moreover, infection with more virulent strains carrying the CagA protein further increases GC risk. 62 Although no direct association between obesity and distal GC has been reported, obesity has been suggested to accelerate the development of H. pylori–associated GC by facilitating cytokine-mediated cross-talk between inflamed gastric and adipose tissues; such cross-talk augments the immune responses in both tissues and thereby promotes the establishment of a pro-tumorigenic gastric microenvironment. 63
Obesity also accelerates gastric carcinogenesis during H. pylori infection by activating signal transducer and activator of transcription-3 (STAT3). 64 STAT3 is a master regulator of inflammatory gene expression that helps create a cancer-promoting microenvironment. 24 In obese individuals, highly expressed leptin and IL-6 activate gastric STAT3. Not surprisingly, high serum IL-6 levels are negatively correlated with GC survival, and disruption of leptin signaling inhibits STAT3-mediated gastric hyperplasia64–66 (Table 1 and Figure 1). By interacting with its transmembrane receptors, leptin activates Janus kinase (JAK)–STAT, mitogen-activated protein kinase (MAPK), phosphatidylinositol 3-kinase (PI3K)–AKT, insulin receptor, and mechanistic target of rapamycin (mTOR) signaling pathways, all of which induce cell proliferation and transformation. 67 STAT3 also promotes the transformation of epithelial cells and is part of the positive feedback loop that underlies the epigenetic changes linking inflammation to cancer. 25 Other pro-inflammatory cytokines such as TNFα, IL-17, and monocyte chemoattractant protein-1 also stimulate the proliferation of human GC cells and inhibit their apoptosis.22,68
The gut microbiota is also an important environmental factor that affects the harvesting and storage of energy from the diet. 69 It is related to the Warburg effect, that is, the aerobic glycolysis that is characteristic of most cancer cells. The oncogenicity of certain pathogens, such as H. pylori, is well established; however, recent studies suggest that commensal bacteria and opportunistic pathogens can also be involved and that infections associated with cancer might be more common than the current estimate of 15%–20%. 70
It is well established that obesity is a state of chronic oxidative stress; methods of improving redox balance include reducing mitochondrial dysfunction and oxidative stress. 71 Excessive levels of reactive oxygen species (ROS) activate multiple protein kinases, including the same protein tyrosine kinases that are overexpressed in GC. 72
CRC
CRC is one of the most prevalent and deadly cancers worldwide; approximately 1 million people are diagnosed with CRC annually, and 60,000 people die of this disease (http://globocan.iarc.fr/). Compared to normal-weight individuals, obese individuals have a 7%–60% higher risk of CRC. 73 Men with a BMI (≥29 kg/m2) have a nearly twofold higher risk of CRC than do those with a BMI (<23 kg/m2) 74 ; women with a BMI ≥29 kg/m2 have a 1.5-fold higher risk. 75 Abdominal adiposity, which is based on waist circumference or the waist-to-hip ratio, is also robustly associated with CRC in both sexes. 1
β-catenin (CTNNB1) is a major component of the WNT/β-catenin signaling pathway, which regulates cell proliferation and is dysregulated in most cancers. 26 A recent study suggests that β-catenin status determines how tumor cells respond to obesity-induced events, and dysregulation of β-catenin signaling has been implicated in CRC development. Obesity and physical inactivity are risk factors for β-catenin-negative CRC but not β-catenin-positive CRC 27 (Table 1).
Accumulating epidemiologic studies challenge the widely accepted notion that diets that are deficient in fiber and other complex carbohydrates are central factors in colorectal carcinogenesis. 76 In obese individuals, systemic chronic low-grade inflammation is largely due to the secretion of hormones, growth factors, and pro-inflammatory cytokines by adipocytes, 77 and long-term inflammation is a risk factor for colitis-associated CRC. 78 Also implicated in CRC development are the insulin/IGF axis and hyperinsulinemia.79,80 In hyperinsulinemic conditions, excess insulin increases the level of bioactive IGF-1 by directly or indirectly decreasing the levels of IGF-binding proteins. 16 When activated, IGF-1 receptors transduce signals that inhibit apoptosis and stimulate cell cycle progression. 17 Moreover, IGF-1 can influence both premalignant and cancerous stages, since both normal and tumorous colorectal epithelial cells express IGF-1 receptors. Taken together, the fact that obesity is a major driver of insulin resistance and hyperinsulinemia suggests that dysregulated insulin and IGF-1 signaling pathways in obese individuals contribute to the development of CRC (Figure 1).
Factors such as adiponectin, leptin, resistin, and ghrelin also promote tumor growth, cell migration, and metastasis.20,21,81–85 During progression from normal colonic mucosa to adenoma to adenocarcinoma, the expression levels of these proteins change, suggesting their involvement in multistep colorectal carcinogenesis.
Various studies have linked obesity to an altered redox state and increased metabolic risk. 86 Notably, oxidative stress is a known etiology for the development of ulcerative colitis and colitis-associated CRC. 87 In fact, recent evidence suggests that oxidative stress may play an important role in all phases of carcinogenesis, including initiation, promotion, and progression. 88
It has been proposed that an unfavorable adipokine profile (i.e. reduced levels of anti-inflammatory and anti-oncogenic adipokines) is a prognostic factor for CRC and that adipokines, their analogs, or their antagonists might be useful in the management and chemoprevention of CRC. 89
Liver cancer
In 2000, liver cancer (LC) was ranked as the fifth and eighth most common malignancy in men and women worldwide, respectively. 90 Most LC patients with early-stage cancers have no obvious symptoms. Consequently, LC is usually diagnosed in its later stages and has a dismal prognosis (the average 5-year survival rate is 6%–11%); it is the third most common cause of cancer-related mortality worldwide. 91 Although hepatitis B and hepatitis C infections are considered major LC risk factors, accumulating evidence suggests that obesity is as well. 92 The risk of LC is 89% higher in obese individuals than in those with normal weight. 93
During hepatocarcinogenesis, β-catenin is frequently mutated and is implicated in the maintenance of tumor-initiating cells, drug resistance, tumor progression, and metastasis.94,95 Moreover, upregulated WNT/β-catenin signaling is believed to increase the likelihood of malignant transformation of preneoplastic adenomas. 96
Liver inflammation has been linked to obesity-induced non-alcoholic fatty liver disease (NAFLD), non-alcoholic steatohepatitis (NASH), fibrosis, and cirrhosis; it also increases the production of cytokines and adipokines that are implicated in hepatocarcinogenesis. Chronic inflammation also increases the abundance of ROS, which induces hepatocellular oxidative stress that causes DNA damage during mitosis and leads to LC development. 97 The main sources of free radicals are hepatocyte mitochondria (which house cytochrome P450 enzymes), endotoxin-activated macrophages (Kupffer cells), and neutrophils. 98 In patients with NAFLD-NASH, the oxidation of excess fatty acids may increase oxidative stress, which triggers pro-inflammatory cytokine release, oncogenic signaling, and epigenetic changes. 99 This sequence of events may occur in the absence of cirrhosis, 100 which is also an oncogenic risk factor. The mechanism of how cirrhosis promotes oncogenesis is unclear; according to one hypothesis, the process of repeated liver cell necrosis and regeneration produces hepatocytes with increased susceptibility to mutagenic agents. 98
Obesity- and inflammation-associated metabolic disorders are characterized by insulin resistance and consequent elevation in the plasma concentrations of insulin, IGF-1, and adipokines. 101 All these factors can increase ROS production. 98 It has been convincingly shown that obesity-induced LC development requires overproduction of the tumor-promoting cytokines IL-6 and TNFα, which induce hepatic inflammation and activation of STAT3. 23 Interestingly, STAT3 was shown to have both tumor-promoting and tumor-suppressing activities in a mouse model of liver tumors. 102 In most conditions, STAT3 acts as an anti-inflammatory agent. 103 However, in liver tumor cells, STAT3 is likely pro-oncogenic (Figure 1).
By inhibiting T-cell function, chronic inflammation suppresses immune surveillance, which is essential for LC development. 104 Severe chronic inflammation can lead to the expansion of liver progenitor cell populations that replace hepatocytes; this expansion may be the precipitating event of LC tumorigenesis. 105 However, it should also be noted that obesity and NAFLD still induced hepatocyte proliferation and decreased apoptosis in a mouse model in which inflammation, fibrosis, and cirrhosis were absent, resulting in hepatic hyperplasia. 106
Pancreatic cancer
Pancreatic cancer (PC) is the eighth leading cause of cancer-related death worldwide, with an incidence of 4.4 per 100,000 in men and 3.3 per 100,000 in women. 107 Despite improvements in surgical techniques and adjuvant medical therapies, the 5-year survival rate of PC has remained only 4% in last four decades; the incidence rate of this disease essentially mirrors its mortality rate. 108 The best means of reducing PC-related mortality is early diagnosis and treatment, ideally during the precancerous stage. Studies have demonstrated that obesity accelerates the development and growth of PC109,110 and that obese individuals have an approximately 20% greater risk of developing PC compared with normal-weight individuals. 111 In both men and women, a BMI ≥25 kg/m2 increases the risk for PC; the risk is especially pronounced in people with a BMI of ≥35 kg/m2. 52
Kirsten rat sarcoma viral oncogene homolog (KRAS) and cyclooxygenase-2 (COX2) are considered key factors in PC initiation and provide mechanistic insight into the connection between obesity and PC development. 28 KRAS, which is a membrane-bound guanine triphosphate (GTP)-binding protein, and prostaglandin E2, whose synthesis is catalyzed by COX2, both inhibit apoptosis and stimulate proliferation and angiogenesis29,30 (Table 1 and Figure 1). Genome-wide association (GWA) studies have identified at least 30 loci that affect the BMI and risk of obesity. 112 Among these loci, the FTO gene was most strongly associated with the BMI and obesity. 113 Moreover, the single nucleotide polymorphism (SNP) rs9939609, which is located in the first intron of FTO, was found to be positively correlated with PC risk in overweight individuals. However, this SNP is also associated with a decreased risk of PC in patients with a BMI <25. 114 A recent GWA study identified frequent SNPs in the NR5A2 gene in PC samples. This gene plays an important role in lipid metabolism and is associated with the BMI.115,116 Polymorphic variations in obesity-related genes may modify the risk of PC individually or jointly, concomitantly with increases in the BMI.
Accumulating evidence implicates 5-lipoxygenase (5-LO) in the growth of pancreatic tumors. Previous studies revealed that 5-LO is overexpressed in tissue samples of primary tumor cells and in established cancer cell lines 117 ; moreover, messenger RNA (mRNA) of the genes that encode 12-LO, 5-LO, and 5-LO-activating protein is overexpressed in adipocytes and adipose tissue of obese Zucker rats compared to their lean counterparts. 118
As mentioned above, obesity is characterized by low-grade chronic inflammation, perhaps due to macrophage infiltration of adipose tissue, which leads to insulin resistance. Alterations in the levels or functions of several obesity-related proteins (e.g. leptin, IGF-1, and peroxisome proliferator–activated receptor-γ) may contribute to PC development by impairing immune function.119–121 Immune dysfunction may result from the production of pro-inflammatory cytokines in macrophages and monocytes, ROS release in neutrophils, and impaired development of natural killer cells owing to the dysregulation of lipid-regulating proteins in obese individuals122–124 (Figure 1). Furthermore, leptin, which is overly abundant in the peripheral tissues of obese individuals, can enhance the invasion of PC cells by increasing matrix metalloproteinase-13 production31,32 (Table 1).
In contrast to the discrepant serum and plasma levels of IGF-1 and IGF-1-binding proteins, epidemiological studies have consistently shown increased glucose levels in PC; this leads to insulin resistance.125,126 In addition, markers of peripheral insulin resistance, rather than those of hyperglycemia or pancreatic β-cell dysfunction, are PC risk factors. 127
Other biological mechanisms, including obesity-related sex steroid effects and obesity-related hypoxia-induced upregulation of vascular endothelial growth factor, may also play a role in tumor progression and growth. Although they are overexpressed during PC progression, their involvement in the mechanism of PC development is unknown.128,129
Interventions aimed at reducing the burden of obesity-associated digestive cancers
It is widely accepted that obesity plays a role in creating a systemic pro-inflammatory environment conducive to the development of diabetes. 130 T2DM can be considered a surrogate risk factor for cancer because it mimics the effects of obesity on metabolism (e.g. it induces insulin resistance and hyperinsulinemia). 131 Glucose-lowering drugs can variably affect circulating insulin levels in T2DM patients. Metformin, the drug of choice for the management of T2DM, 132 reduces levels of both circulating glucose and insulin in patients with insulin resistance and hyperinsulinemia. By activating the liver kinase B1/AMP-activated protein kinase (LKB1/AMPK) pathway, metformin inhibits mTOR to negatively modulate protein synthesis in cancer cells. 133 T2DM patients receiving biguanide metformin have a lower incidence of cancer than do those receiving other medications such as insulin and sulfonylureas.134,135
Physical activity may counteract, at least in part, the oncogenic effects of obesity and reduce the likelihood of developing cancer. 136 Importantly, physical activity may modify genetic susceptibility to obesity and decrease the genetic burden, which is higher in physically inactive versus active people.137,138 A previous report suggests that obese individuals are less likely to engage in outdoor physical activity, which limits their exposure to the sun and hence their production of vitamin D. However, it is unlikely that vitamin D deficiency mediates the effects of obesity on cancer development. 139 Serum levels of C-reactive protein, a marker of inflammation, are higher in overweight and obese individuals than in normal-weight individuals, and decline with weight loss. 140 Inflammation is a common element in the pathogenesis of both obesity and cancer, and mitigation of inflammation via weight loss may therefore decrease the risk of cancer.13,14
Reduced or modified dietary fat intake and calorie restriction are thought to prevent obesity-associated cancer development.141,142 High fat and carbohydrate levels may increase the production of ROS by saturating the electron transport chain; the consequent oxidative stress may cause chronic inflammation concomitantly with the development of obesity. 143 Diet modulation has been shown to be an effective means of preventing and treating breast and lung cancer metastases. 144 Moreover, calorie restriction reduces insulin and IGF-1 levels, and consequently decreases chronic inflammation. On the contrary, adiponectin levels, which are usually low in obese individuals, ought to be increased to counteract obesity-induced metabolic perturbations in calorie-restricted conditions. 145 An increase of the adiponectin has been correlated to the improvement of whole-body energy metabolism and of adipose tissue functions.146,147
Conclusion
The prevalence of obesity is increasing in developed countries, in which 60%–70% of the adult population is currently overweight or obese. Although it is widely accepted that obesity increases the incidence and mortality of multiple cancers, the molecular mechanisms of how this occurs are unclear. Herein, we have provided a detailed overview of obesity and its relationship to cancers of the digestive system. Factors clearly associated with these cancers include obesity-associated gene mutations, insulin and IGF signaling pathways, chronic inflammation, and altered adipokine levels. These factors usually act interdependently rather than independently in carcinogenesis. Optional interventions such as physical activity, diet modulation, and calorie restriction may reduce the burden of obesity-associated digestive cancers.
Footnotes
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the National Natural Science Foundation of China (Grant Numbers 31301068 and 81370165), the Natural Science Foundation of Zhejiang Province (No. LY17C060002), the Scientific Innovation Team Project of Ningbo (No. 2014B82002), and the K. C. Wong Magna Fund in Ningbo University.