
Abstract
Many cancers might be influenced by obesity, including breast cancer, the leading cause of cancer death among women. Obesity is a complex state associated with multiple physiological and molecular changes capable of modulating the behavior of breast tumor cells and the surrounding microenvironment. This review discussed the inverse association between obesity and breast cancer among premenopausal breast cancer females and the positive association among postmenopausal. Four mechanisms may link obesity and breast cancer including leptin and leptin receptor expression, adipose chronic inflammation, sex hormone alternation, and insulin and insulinlike growth factor 1 (IGF-1) signaling. Leptin has been involved in breast cancer initiation, development, and progression through signaling transduction network. Leptin functions are strengthened through cross talk with multiple oncogenes, cytokines, and growth factors. Adipose chronic inflammation promotes cancer growth and angiogenesis and modifies the immune responses. A pro-inflammatory microenvironment at tumor site promotes cytokines and pro-inflammatory mediators adjacent to the tumor. Leptin stimulates pro-inflammatory cytokines and promotes T-helper 1 responses. Obesity is common of chronic inflammation. In obese patients, white adipose tissue (WAT) will promote pro-inflammatory mediators that will encourage tumor growth and WAT inflammation. Sex hormone alternation of estrogens is associated with increased risk for hormone-sensitive breast cancers. Estrogens cause tumorigenesis by its effect on signaling pathways that lead to DNA damage, stimulation angiogenesis, mutagenesis, and cell proliferation. In postmenopausal females, and due to termination of ovarian function, estrogens were produced extra gonadally, mainly in peripheral adipose tissues where adrenal-produced androgen precursors are converted to estrogens. Active estradiol leads to breast cancer development by binding to ERα, which is modified by receptor’s interaction of various signal transduction pathways. Hyperinsulinemia and IGF-1 activate the MAPK and PI3K pathways, leading to cancer-promoting effects. Cross talk between insulin/IGF and estrogen signaling pathways promotes hormone-sensitive breast cancer development. Hyperinsulinemia is a risk factor for breast cancer that explains the obesity-breast cancer association. Controlling IGF-1 level and targeting IGF-1 receptors among different breast cancer subtypes may be useful for breast cancer treatment. This review discussed several leptin signaling pathways, highlighting the potential advantage of targeting leptin as a potential target of the novel therapeutic strategies for breast cancer treatment.
Keywords
Traditionally, obesity was defined as an increase in body weight that was greater than 20% of an individual’s ideal body weight, and overweight was defined as a 15% to 20% increase over ideal body weight. The World Health Organization and the National Institutes of Health define obesity as a body mass index (BMI, defined as weight (kg)/height (m2)) greater than 30 kg/m2 (30.0-34.9, grade I; 35.0-39.9, grade II; and grade III equal or greater than 40) and overweight as a BMI greater than 25 kg/m2. The World Health Organization considered global obesity an epidemic, in which more than 1.9 billion adults worldwide were overweight and 650 million (13% of the world’s adult population) were obese. 1
Obesity prevalence is varied with age, sex, race, and across and within countries. 2 Considerable racial differences existed, with obesity rates ranging from 2.6% for Chinese and 3.3% for Vietnamese women to 34.9% for American Indians/Alaska Natives and 60.2% for Samoans. 3 Unfortunately, transition in nutrition, economic growth, and technological changes, increasing the prevalence of obesity alarmingly. Researchers have found that over the past 3 decades, there is a startling increase in obesity, with no decline in any country globally. 4
Obesity is associated with increased risk of many chronic conditions such as diabetes, heart disease, and hypertension and increased risk of cancer. Abdominal adiposity plays a role in generating an environment that enriches cancer development. 5 The link between obesity and cancer, especially breast, endometrial, ovarian, thyroid, and prostate cancers, was reported. 6 This link built by many risk factors including hyperinsulinemia, inflammation, and adipokines secretion will be discussed in more detail.
Breast cancer is universal among women and is the leading cause of cancer-related mortality worldwide. 7 Breast cancer makes up 25% of all new cancer diagnoses in women globally. 8 Although age, genetics, family history,9,10 and reproductive factors are considered as well-known risk factors for breast cancer incidence,11,12 alcohol drinking, smoking, excess body weight, vitamin D,13-15 and physical inactivity may be considered as modifiable risk factors (represent 20% of breast cancers risk factors). However, to reduce breast cancer incidence, many lifestyle changes are necessary. The effect of obesity on developing breast cancer has been evaluated among different population groups, showing differences across pre- and postmenopausal females. 16
The association between obesity and breast cancer as a risk factor among premenopausal women is still debatable,16,17 which may be due to the differences in ethnicities and hormonal receptor status.16,17 However, the American Institute for Cancer Research highlighted that being overweight/obese decreased the risk of premenopausal breast cancer. 18 An inverse association between premenopausal breast cancer risk and BMI was reported in a pooled study that enrolled 337 819 women and 4385 invasive breast cancers. The inverse association was confirmed when comparing women who had a BMI >31 kg/m2 versus those who had a BMI ⩽21 kg/m2. 19 Another meta-analysis enrolled more than 2.5 million women and 7930 premenopausal breast cancers which demonstrated that premenopausal breast cancer risk is reduced by approximately 8% per 5 kg/m2 BMI increase. 20 This inverse association can be explained pathophysiologically by a negative feedback mechanism on the hypothalamic pituitary axis that reduces gonadotrophin release from high estrogens produced from ovary and peripheral tissue (Figure 1). 21

Diagrammatic representation of the origins, target organs, and feedback mechanisms of the hormones involved in the hypothalamic-pituitary-ovarian axis. FSH indicates follicle-stimulating hormone; GnRH, gonadotropin-releasing hormone; LD, luteinizing hormone.
Obesity is associated with increased breast cancer risk among postmenopausal women. Clinical trial of 67 142 postmenopausal women showed that obese postmenopausal women are at greater risk of developing breast cancer compared with their normal-weight equivalents (BMI >35.0 kg/m2). 22 This increase was also demonstrated in a population-based cohort study of 5.24 million 23 and confirmed in a meta-analysis of that enrolled 89 epidemiologic reports. 24 This association was limited for estrogen and progesterone receptor–positive postmenopausal breast cancer. The relative risk increased by approximately 40% for receptor-positive postmenopausal breast cancer. 24 Among postmenopausal nonusers of hormonal therapy, higher BMI was significantly associated with an increased risk of less aggressive ER+, PR+, HER2−, Bcl-2+, and p53− tumors. 25 These less aggressive molecular subtypes of breast cancer are characterized by a lower proliferation rate (Ki67low), Bcl-2 positivity, and p53 negativity—immunohistochemical characteristics that are each associated with better prognosis. 26
Obesity-Breast Cancer Molecular Mechanism
The molecular mechanisms that link obesity-breast cancer are complex, but 4 mechanisms can explain this link: leptin and leptin receptor expression, adipose chronic inflammation, sex hormone alternation, insulin, and insulinlike growth factor 1 (IGF-I) signaling.
Leptin and leptin receptor expression
Leptin is the most important mediator for obesity-breast cancer link that promotes tumor initiation, development, growth, and metastasis. 27 Leptin increases breast cancer risk by cross talk with other signaling molecules, including ERα, growth factors, Notch, and inflammatory factors. 28 Cross talk between leptin and IGF-I signaling promotes invasion of breast cancer cells via transactivation of epidermal growth factor receptor (EGFR). 29 Novel signaling cross talk between leptin, Notch, and interleukin 1 (NILCO) develops leptin-induced cancer. Leptin-NILCO signaling mediates the activation of cancer stem cells (CSCs). 30 Cancer stem cell signaling promotes leptin receptor expression, by activation of STAT3 pathway and increase in expression of stem cell markers such as OCT4 and SOX2. 31 The interaction between stromal cells (cancer-associated fibroblasts and breast adipocytes) and breast CSCs, leptin, and its receptor offers novel insights for realizing how breast cancer tumor microenvironment promotes cancer growth and development.
Leptin-induced epithelial-mesenchymal transition (EMT) that is associated with morphological changes by decreasing cell to cell contact and promoting elongated morphological shape. Epithelial-mesenchymal transition is defined as the ability of epithelial cells to convert from a polarized morphology to a loose mesenchymal phenotype that stimulates cancer metastases. 32 The mechanism of this transition is by reducing the expression levels of epithelial phenotype markers E-cadherin and keratin, increasing the expression of mesenchymal phenotype marker vimentin, and raising the expression of EMT-induced transcription factor ZEB-1. 33 Epithelial-mesenchymal transition encourages the transient cells to increase cell mobility, tumor invasion and metastasis, and to be more resistant to cytotoxic drugs. 34
Adipose inflammation
Adipose chronic inflammation promotes cancer growth, encourages angiogenesis, and modifies antitumor immune responses. 35 A pro-inflammatory microenvironment at tumor site is generated from inflammatory points known as crown-like structures (CLS), a histologic biomarker of inflammation. 36 These will promote cytokines and pro-inflammatory mediators, such as cyclooxygenase-2 (COX-2), TNF-α, IL-6, monocyte chemoattractant protein-1 (MCP-1), and interleukin (IL)-1β 37 adjacent to the tumor. Leptin stimulates pro-inflammatory cytokines and promotes T-helper 1 responses. 38 Obesity is common of chronic inflammation. In obese patients, white adipose tissue (WAT) will promote pro-inflammatory mediators that will encourage tumor growth, and WAT inflammation is shared in obese and overweight individuals. 37
Inflammasome is another important source of inflammation in adipose tissue among obese individuals. Inflammasome activates the maturation of pro-inflammatory cytokines, including IL-1β and IL-18. 39 Adipocytes express multiple inflammasome-related genes, including PYCARD, CASP1, NLRP3, IL-1β, and TNF, 40 and these are common in obese subjects. Activated inflammasomes promote inflammatory microenvironment that promotes tumor growth and metastasis. 40 NLRP3 inflammasome interferes with apoptosis and cell cycle, 41 whereas NLRC4 inflammasome/IL-1β increases angiogenesis through an activation of vascular endothelial growth factor A in adipocytes. 42 Recently, Raut et al 41 showed that activation of inflammasomes encouraged leptin-induced growth of breast cancer cells by mediating estrogen receptor signaling and reactive oxygen species (ROS) production. Thus, the complicated function of inflammasomes raises new challenges for novel approaches that can be used for treatment of obese patients with breast cancer.
Sex hormone alternation
Circulating estrogens have been associated with increased risk of hormone-sensitive breast cancers, 43 mainly among postmenopausal females. The mechanism of how estrogens cause tumorigenesis is by its effect on signaling pathways that lead to DNA damage, stimulation angiogenesis, mutagenesis, and cell proliferation. 44 Meta-analyses showed that the estrogenic background among postmenopausal obese is an important risk factor for breast cancer with estrogen receptor–positive individuals.45,46 In postmenopausal females and due to termination of ovarian function, estrogens produced were extra gonadally, mainly in peripheral adipose tissues where adrenal-produced androgen precursors are converted to estrogens. This process was catalyzed by the cytochrome P450 enzyme aromatase. 20 This leads to excess adipose tissue which causes increases in estrone and estradiol, and free estradiol levels were linked with high BMI among postmenopausal women.47,48 Estrogen production by aromatase conversion from androgens plays an important role in breast tumor promotion. 49 Active estradiol leads to breast cancer development by binding to ERα, which is modified by receptor’s interaction with various signal transduction pathways. 50 Developing selective estrogen receptor modulators, aromatase and sulphatase inhibitors, that block the cross talk in the signal transduction pathways could lead to novel treatment strategies for breast cancer.
Insulin and insulin growth factor
Insulin and hyperinsulinemia are signs for obesity, and hyperinsulinemia is associated with increased IGF-1. 51 IGF-1 is a hormone that exhibits mitogenic and antiapoptotic properties. 52 IGF has a multifactorial role in cancer, and data suggest a slight increased risk of some cancers due to higher activity of the IGF system. On the contrary, patients with congenital deficiencies in IGF-1 have a protective effect against developing cancer. 53 There is a bidirectional relationship between dysregulated/imbalanced glucose/insulin metabolism and breast cancer among overweight/obese females. 54 Hyperinsulinemia is accompanied with increased synthesis of IGF-1 which activates the MAPK and PI3K pathways, leading to cancer-promoting effects of obesity. 51 Cross talk between insulin/IGF and estrogen signaling pathways promotes hormone-sensitive breast cancer development. 55 Hyperinsulinemia is an independent risk factor for breast cancer and may explain the obesity-breast cancer association. 56 In vitro and in vivo studies showed that IGF-I signaling cascade mediates breast cancer growth, migration, angiogenesis, and survival in breast cancer models.57,58
Hyperinsulinemia is common in obese women. 59 Insulin stimulates cell proliferation in human breast cancer cell lines 60 and alters estrogen levels 61 which increase breast cancer risk. Pooled data analysis of 17 studies has shown a significant association between IGF-I levels and breast cancer risk. 62 Increased insulin and IGF-1 levels were associated increased mortality, 63 whereas activated IGF-I receptors were associated with poor survival patients with breast cancer. 64 Controlling IGF-1 level and targeting IGF-1 receptors among different breast cancer subtypes may be useful for breast cancer treatment among specific subgroup. 58
Leptin Signaling Pathways in Breast Cancer
Signaling pathways that mediate the leptin effects on breast cancer cells include JAK/STAT, MAPK, and PI3K pathways (Figure 2):
JAK/STAT signaling pathway. Through activating the JAK2/STAT3 pathway, leptin promotes the progression of breast cancer in the cancer cells. By binding of leptin to its receptor, JAK2 kinase is activated. This provides an anchoring site for the STAT3 protein. Once STAT3 is bound to the receptor, it is phosphorylated by JAK2 and STAT3 proteins dimerize, which is then translocated into the nucleus. These activate the transcription of several genes that involve in cell proliferation such as c-myc, cyclin D1, p21/waf1, c-jun, junB, erg-1, and Bcl-2. 66
MAPK pathway. Activating the MAPK signaling pathway plays an important role in the activation of ERK 1/2, p38, and JNK. 67 Activating MAPK pathway induces the activation of transcription factors, such as c-jun, c-fos, c-myc, and erg-1, which regulate cell proliferation. Leptin promoting growth via ERK pathway has been demonstrated in breast cancer models.
PI3K pathway. This is an important link between obesity, leptin, and increased risk of breast cancer. 68 It affects cellular properties more rapidly than other pathways. It acts through protein phosphorylation of insulin receptor substrate that controls the PI3K pathway and regulates Akt signaling. 69 Leptin enhances the proliferation, migration, and invasion of breast cancer cells via acetyl-CoA acetyltransferase 2 (ACAT2) upregulation through the PI3K/AKT/SREBP2 signaling pathway. 70
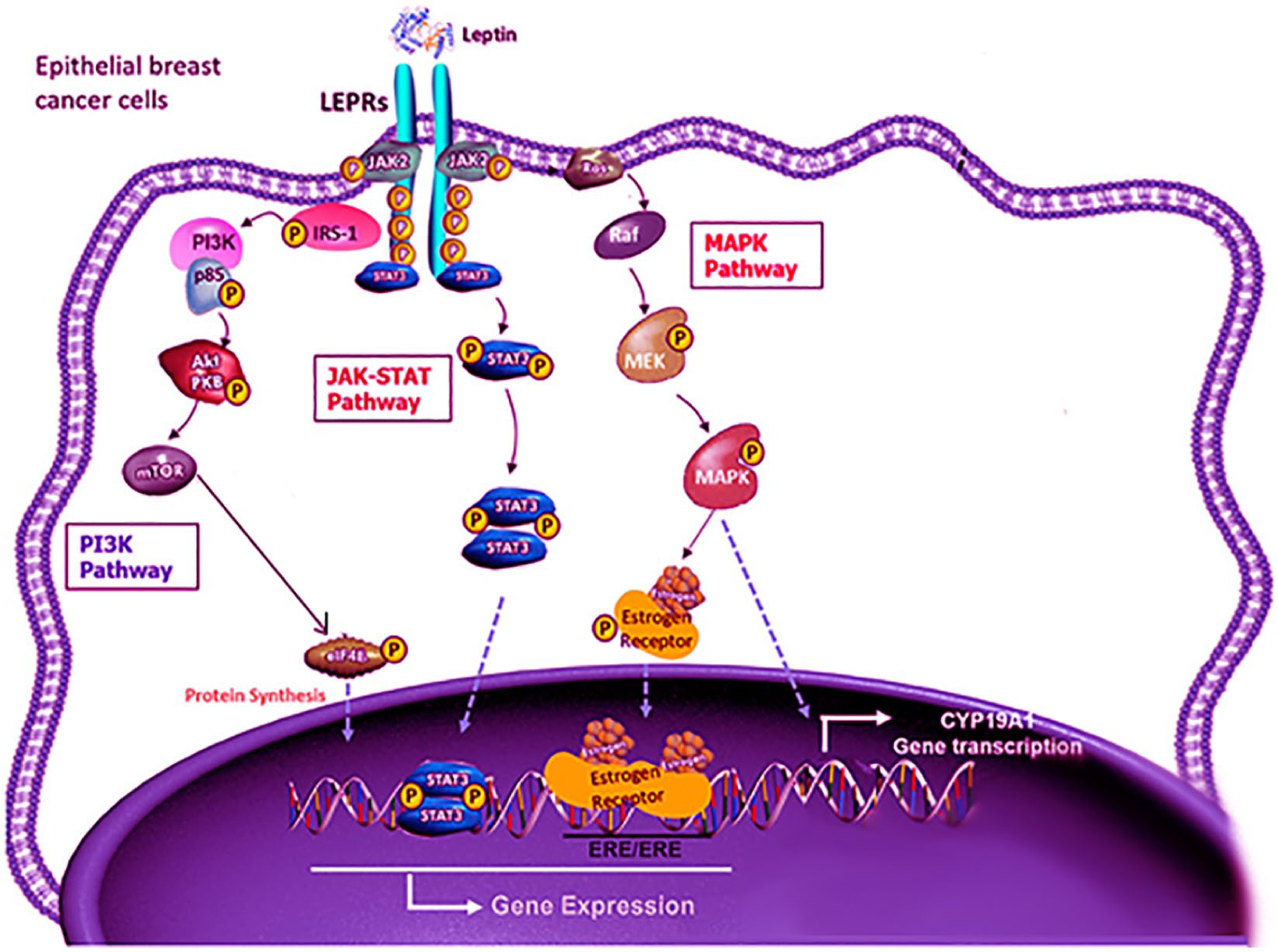
Signaling pathways that mediate the leptin effects on breast cancer cells include JAK/STAT, MAPK, and PI3K pathways.
The link between obesity and leptin in breast cancer was studied broadly in vivo. Zucker rats that lack leptin response due to leptin receptor missense mutation are models that reproduce obese metabolic syndrome. Administration of a carcinogen in Zucker rats results in breast carcinoma development in obese Zucker rats compared with lean controls. 71 Other genetic models of obesity, including mouse mammary tumor virus (MMTV)-TGF-α/Lep(ob)/(ob) (leptin-deficient) and MMTV-TGF-α/Lepr(db)/(db) (leptin receptor-deficient) mice, did not develop mammary tumors compared with wild-type mice.72,73 But these mouse models showed defective mammary gland development and demonstrate a bias for studying the specific involvement of leptin in obese-induced cancers. So, rebuilding of leptin receptor signaling in the brain of db/db mice restored the development of the mammary gland. 74 Mice deficient in peripheral leptin receptor and with an intact central leptin signaling showed a decreased mammary tumor growth. Leptin promotes tumor progression in MMTV-Wnt-1 mice, whereas mammary tumor growth was inhibited in leptin-deficient mice (Lepob/ob). 75 Recently, knockdown of leptin in adipose stromal/stem cells isolated from obese patients resulted in reduced tumor growth and metastasis in severe combined immunodeficiency/beige mice. 76
The Link Between Leptin and Breast Cancer
Leptin is a hormone that is produced by adipose tissue (normal and malignant tissue) and overexpressed in obese and overweight people. 77 It is encoded by the obese (Ob) gene that keeps energy homeostasis, through a central feedback mechanism at the hypothalamus level. It controls adipose tissue growth and cell proliferation including breast tissue. 77 Leptin levels were related to breast cancer aggressiveness and can predict the type, grade, stage, lymph node involvement, hormone receptors, and recurrence in breast cancer. 78 Leptin affects breast cancer biology in an endocrine, paracrine, and autocrine manner.79,80 Leptin actions were through leptin receptor (ObR) that is encoded by db gene. 79 Binding leptin to ObR activates JAK2/STAT3, MAPK, and PI3K/Akt signaling pathways that regulate cell proliferation.
Leptin promotes breast cancer progression due to its role in EMT. Epithelial–mesenchymal transition releases epithelial cells from the surrounding tissue and rearranged the cytoskeleton, permitting the movement of epithelial cells into the extracellular matrix. 81 In addition, leptin involved in breast cancer metastasis. 82 Females with elevated serum leptin have higher breast cancer risk.83,84 A meta-analysis of 43 studies suggests that the serum leptin may provoke a major role in the pathogenesis and metastasis of breast cancer. 78 Another meta-analysis of 23 reports 85 showed that circulating leptin levels among healthy people were less than patients with breast benign disease, less than patients with breast cancer, and less than lymph node metastasis-positive patients, signifying leptin levels as a suitable diagnostic tool for neoplasia. A third meta-analysis of 35 studies suggested that leptin could be a potential biomarker for breast cancer risk in women, especially overweight/obese or postmenopausal women, and may be useful biomarker for preventive treatments through identifying subjects with a high risk for breast cancer. 86 Moreover, leptin receptor mRNA expression in breast cancer tissue predicts poor prognosis for patients with high serum leptin levels. 87 These findings may highlight the potential advantage of targeting leptin signaling to block breast cancer malignancy.
Leptin signaling control many molecules involved in cell proliferation, adhesion, invasion, migration, inflammation, and angiogenesis. These molecules implicated in breast carcinogenesis with the regulation of the expression of VEGF,29,88 E-cadherin, 89 and cyclin D1. 29 In breast cancer, leptin has roles in intensifying the activity of signaling pathways involved in the cell proliferation and roles in downregulation of the apoptotic response. 67 It also activates ROS production in human epithelial mammary cells. 90 Leptin also regulates metabolic reprogramming which promotes cellular growth. 91 All these will modify the microenvironment at adipose tissue that will promote breast cancer development.
Leptin is associated with mammary tumorigenesis by guiding cell-to-cell communication 92 by regulating exosome biogenesis and release in ERα-positive MCF-7 and triple-negative MDA-MB-231 breast cancer cells. Leptin action increased the Tsg101 protein that interacts with chaperone protein Hsp90. 92 Obstruct cell-to-cell communication and damage in exosome secretion might be a novel therapeutic strategy in breast cancer treatment.
Leptin Cross Talk in Breast Cancer
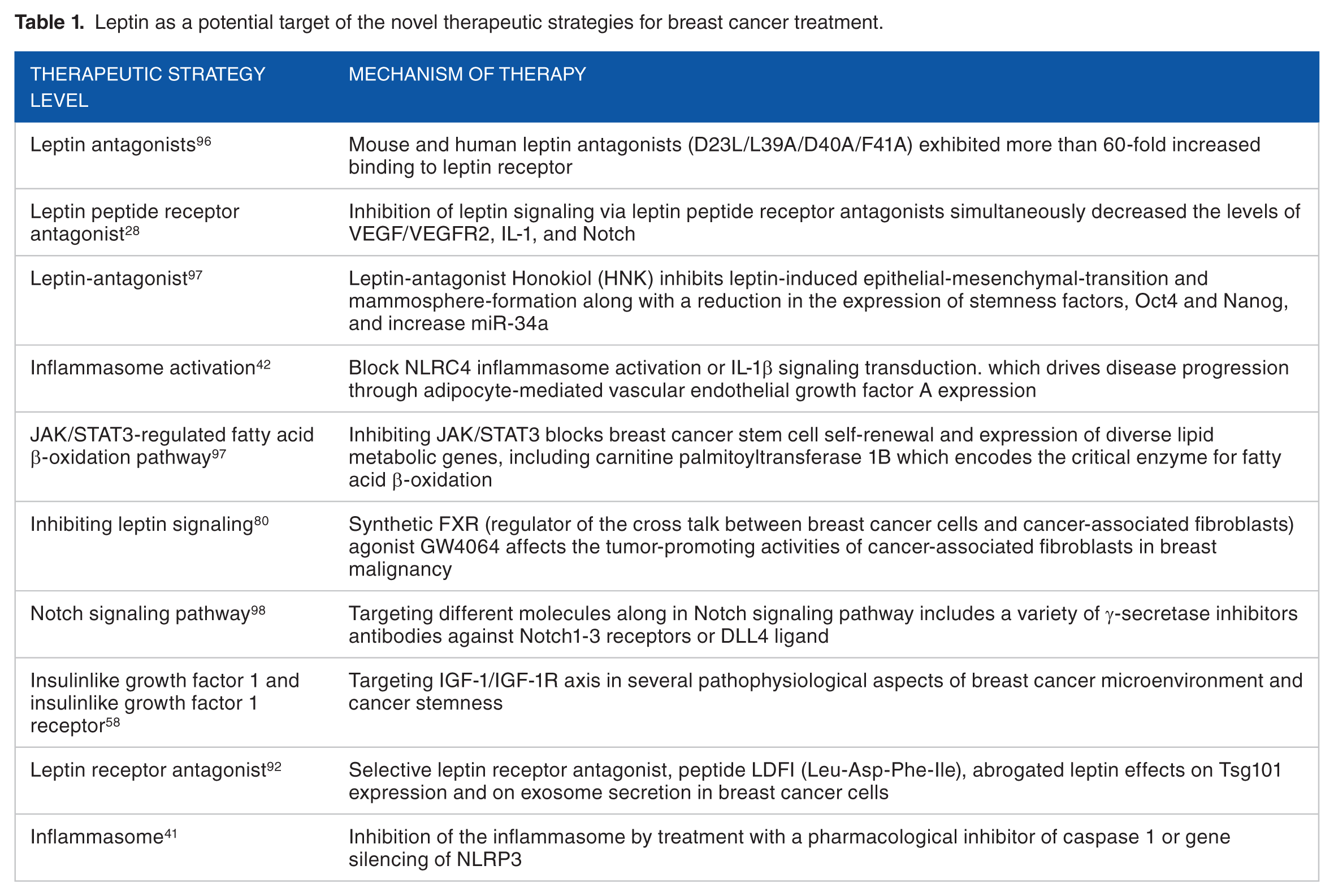
Leptin has interrelated breast cancer initiation, development, and progression through signaling transduction network. Leptin functions are strengthened through cross talk with multiple oncogenes, cytokines, and growth factors. 93 Cross talk between leptin, IL-1, and NILCO found in breast cancer cells links pro-inflammatory and pro-angiogenic signals that induce breast cancer. 28 Furthermore, leptin increases the cross talk between breast cancer cells and tumor-associated macrophages M2 by stimulating IL-18 and IL-8 production that promotes tumor growth. 94 In addition, leptin may stimulate CSCs enrichment and EMT phenotype by upregulating the expression CSC/EMT-related genes. 95 Furthermore, activation of HIF-1α and NF-κB by leptin regulates VEGF and promotes angiogenesis in mammary tumors. 88 Also, cross talk between leptin and IGF-1 signaling promotes EGFR that stimulates proliferation of triple-negative breast cancer cells. 29 Leptin can stimulate HER2 through the activation of the EGFR and the activation of JAK2, resulting in breast cancer cell growth. 65 Studying signaling transduction networks and leptin cross talk in breast cancer may guide the identification of novel therapies to reduce breast cancer initiation, development, and metastasis. So several therapeutic approaches have been suggested that may interfere and prevent leptin-related breast cancer, which is summarized in Table 1. These studies address novel strategies for leptin obesity–related breast cancer prevention, using approaches directed against breast cancer development. These mechanisms include leptin antagonists, leptin peptide receptor antagonist, inflammasome activation, JAK/STAT3-regulated fatty acid β-oxidation pathway, inhibiting leptin signaling, Notch signaling pathway, and IGF-I and IGF-I receptor. But, the main concern is the correct selection of at-risk populations (the baseline of adipokine status, BMI, insulin-IGF-1 level, WAT inflammation, sex hormone level, and type of sex hormone receptors). Regarding the growing knowledge on leptin mechanisms in breast cancer carcinogenesis, the ongoing development of targeting leptin signaling mechanisms will open new promising approaches for breast cancer prevention and avoiding treatment resistance.
Leptin as a potential target of the novel therapeutic strategies for breast cancer treatment.
Conclusions
Many mechanisms link leptin obesity-breast cancer, including serum leptin level and leptin receptors expression, chronic adipose inflammation, sex hormone alternations, and insulin-IGF-1. Signaling pathways that mediate the leptin effects on breast cancer cells include JAK/STAT, MAPK and PI3K pathways were discussed. And in vivo studies of the role of leptin in different models were discussed including Zucker rats, mammary tumor virus (MMTV)-TGF-α/Lep(ob)/(ob) (leptin-deficient), MMTV-TGF-α/Lepr(db)/(db) (leptin receptor–deficient) mice, and MMTV-Wnt-1 mice models. Several therapeutic approaches were suggested including leptin and leptin receptor antagonists, signaling pathways, inflammasome targeting, Notch signaling, and IGF-I and IGF-I receptor targeting.
Footnotes
Acknowledgements
The authors thank the Hashemite University for their continuous support.
Declaration of conflicting interests:
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding:
The author(s) received no financial support for the research, authorship, and/or publication of this article.