
Abstract
Exosomes secreted from the cell to the extracellular environment play an important role in intercellular communication. Next-generation sequencing technology, which has achieved great development recently, allows us to detect more complete data and gain even deeper analyses of RNA transcriptomes. In our research, we extracted exosomes from different gastric cancer cell lines and immortalized normal gastric mucosal epithelial cell line and examined the amounts of exosomal proteins and RNAs. Our data showed that the secreted amount of cancer cell–derived exosomes, which contain proteins and RNAs, was much higher than that of normal cell–derived exosomes. Moreover, next-generation sequencing technology confirmed the presence of small non-coding RNAs in exosomes. Based on publicly available databases, we classified small non-coding RNAs. According to the microRNA profiles of exosomes, hsa-miR-21-5p and hsa-miR-30a-5p were two of the most abundant sequences among all libraries. The expression levels of the two microRNAs, miR-100 and miR-148a, in exosomes were validated through reverse transcription polymerase chain reaction. The reverse transcription polymerase chain reaction result, consistent with the trend of sequencing result, indicated a significant difference in exosomes between gastric cancer and gastric mucosal epithelial cell lines. We also predicted novel microRNA candidates but they need to be validated. This research provided an atlas of small non-coding RNA in exosomes and may make a little contribution to the understanding of exosomal RNA composition and finding parts of differential expression of RNAs in exosomes.
Introduction
Exosomes are a class of extracellular vesicles (EVs) that are secreted outside the cells. They have received much attention in recent years. It is commonly reported that exosomes are nanoscale vesicles, with 40–100 nm diameter and 1.10–1.21 g/mL density.1–3 At first, the inward budding of late endosomes formed multivesicular endosomes, which then fused with the outer cell membrane, and finally, the exosomes packaged in multivesicular endosomes are released outside. 4 It has been widely accepted that exosomes can exist in many kinds of body fluids, physiologically and pathologically, including plasma, urine, saliva, and malignant effusions. 5 Studies reported that exosomes can be found in the culture supernatant of many different types of cells, such as normal cells (e.g. hemopoietic stem cells, dendritic cells, epithelial cells) or tumor cells (e.g. gastric cancer (GC) cells and pancreatic cancer cells). 6 Exosomes contain various proteins, messenger RNA (mRNA), and microRNA (miRNA), depending on the cellular origin. These compositions in exosomes can be transferred from donor cells into neighbor or distant cells and then produce biological effects in the target cells.1,5,6 These diverse biological functions that exosomes mediated may include cell proliferation, invasion and metastasis, angiogenesis, immune response, and so on.
In particular, exosomes produced by cancer cells have a very important effect on metastasis by modifying the tumor microenvironment. Many studies have shown that exosomes derived from cancer cells may trigger the differentiation of normal stromal fibroblasts to myofibroblasts7,8 or the differentiation of macrophages to tumor-associated macrophages.9,10
As RNA is the main component of exosomes, we wonder whether the differences in RNAs in different kinds of exosomes can produce changes. We hypothesize that different exosomes are composed of different RNAs and finally affect the tumor microenvironment.
miRNAs are one class of small non-coding RNAs (ncRNAs) that are generally 18–24 nucleotides in length. miRNAs can suppress the expression of many key proteins by base pairing with complementary sequences of the mRNAs. Dysregulated miRNAs play important roles, such as regulating cell proliferation, apoptosis, and invasiveness in various cancers. 11 Since exosomes play an important role in transferring cellular genetic materials, we focus more on the miRNAs present in the exosomes. However, both the distribution characteristics and the profiles of miRNAs expression in exosomes remain unknown.
In our study, next-generation RNA sequencing (NGS) technology was utilized. This technology is commonly used in various biological applications in recent years. It can detect all RNA subtypes precisely and sensitively, especially including unannotated or low-abundance sequencing fragments.
We extracted exosomes derived from cell lines: four human GC cell lines and an immortalized gastric mucosal epithelial cell line (GES-1). GES-1 is the only cell line established from normal gastric epithelium cells. Considering that fibroblast cells, endothelial cells, or other normal cells have huge differences compared with gastric tissue, we exclude them out.
In addition, we used NGS to get sequencing data of small ncRNAs in different exosomes. These sequencing data were analyzed by bioinformatics method. Our research provided a new insight into an atlas of small ncRNAs in exosomes and may make a little contribution to the understanding of exosomal RNA composition and find parts of differential expression of RNAs in exosomes.
Materials and methods
GC cell lines and cell culture
The human GC cell lines, MKN45, SGC7901, NCI-N87, and AGS, and a normal cell line, GES-1, were preserved in our laboratory. The cells were repeatedly cultured in RPMI 1640 (Genom Biotechnology, Hangzhou, China) supplemented with 10% fetal bovine serum (FBS; Thermo Fischer Scientific, San Jose, CA, USA), 100 U/mL penicillin, and 100 µg/mL streptomycin in a humidified cell incubator with an atmosphere of 5% CO2 at 37°C. Cells growing at an exponential rate were used for the experiments.
Generation of exosome-depleted FBS and isolation of exosomes
FBS (Thermo Fischer Scientific) depleted of exosomes (exosome-depleted FBS), which was used to prevent from being contaminated by exosomes in FBS, was generated by ultracentrifugation at 110,000g at 4°C for 4 h, and careful collection and filtration at 0.22 µm of the resulting supernatant. Cells were cultured in culture media with exosome-depleted FBS for 48 h at 80%–90% confluence. The number of cells was about the same, nearly 1.2 × 107 cells in one dish. Conditioned media was harvested and centrifuged at 200g at 4°C for 5 min and then at 2000g at 4°C for 30 min to precipitate cell debris. The procedure of cell culture and collection medium among different types of cell lines was standardized. The supernatant was ultracentrifuged at 110,000g at 4°C for 70 min to pellet the exosomes. Exosomes were isolated from cell culture media with Total Exosome Isolation Reagent (Life Technologies, Carlsbad, CA, USA) according to the manufacturer’s instructions. Briefly, cell culture media was mixed with the Isolation Reagent at a ratio of 2:1 and incubated overnight at 4°C. After centrifugation at 10,000g for 70 min, exosome pellets were precipitated. Exosomes were resuspended in either phosphate-buffered saline (PBS) for electron microscopy analyses or lysis buffer for Western blot analyses.
Transmission electron microscopy
Negative staining was used for transmission electron microscopy (TEM) examination of exosomes. For negative staining, the exosome pellet was resuspended in PBS. Then, the resuspending solution was loaded onto formvar/carbon-coated EM grids, fixed with 2% paraformaldehyde (PFA), and stained with 1% aqueous uranyl acetate, then dried at room temperature. The samples were examined using an FEI Tecnai G2 Spirit device TEM (FEI, Hillsboro, OR, USA).
Western blot analyses
Exosome pellets were lysed using radioimmunoprecipitation assay (RIPA) buffer (Thermo Fischer Scientific). The protease inhibitor phenylmethylsulfonyl fluoride (PMSF; Thermo Fischer Scientific) was also added in the lysate solution. The protein concentration of the lysates was measured by the BCA Protein Assay Kit (Thermo Fischer Scientific). Equivalent amounts of protein were mixed with 5 × Lane Marker Non-Reducing Sample Buffer (Thermo Fischer Scientific) and β-mercaptoethanol (20:1) and boiled for 10 min. Then, the mixture was subjected to 10% polyacrylamide gel electrophoresis and transferred onto polyvinylidene difluoride (PVDF) membranes (Merck Millipore, Bedford, MA, USA). The membranes were blocked with 5% non-fat milk in Tris-buffered saline for 2 h at room temperature and subsequently incubated with the CD36 (1:500 dilution; Abcam, Cambridge, MA, USA) and CD9 antibodies (1:500 dilution; Abcam) overnight at 4°C. Membranes were then washed thrice with 0.1% Tween 20 in PBS and incubated with the horseradish peroxidase–conjugated secondary antibody followed by visualization with enhanced chemiluminescence. There is no protein generally accepted as the loading control for exosomes at present.
Isolation of total RNA
TRIzol (Life Technologies) was used to extract total RNA from exosomes. After the extraction, we immediately measured it by a NanoDrop Spectrophotometer and an Agilent 2100 Bioanalyzer. The total RNA was sequenced or reverse transcribed for further validation.
Construction of small RNA libraries and deep sequencing
Sequencing of samples was conducted by a commercial service (Ruibo, Guangzhou, China) using an Illumina HiSeq™ 2500 device. A volume of 40 µg total RNA from exosomes was used to obtain 18–30 nt small RNA using acrylamide gel purification method. The small RNA was decapped with Tobacco Acid Pyrophosphatase (TAP) treatment, separately ligated to identical bar-coding 5′- and 3′- adaptors, acrylamide gel purified, reverse transcribed, and polymerase chain reaction (PCR)-amplified. Then the original sequence data were sequenced in the type of single end (SE): 1 × 50 bp, and retrieved by trimmed inserts, finally saved as FASTA files. The results have been submitted to the National Center for Biotechnology Information (NCBI) Sequence Read Archive (SRA) under accession number SRP070915.
Bioinformatics analysis
The Illumina read libraries regarded as raw data were converted into FASTA file format. The 3′ adapter sequences and 5′ adapter contaminants were trimmed. Low-quality reads and too short reads were discarded. Then, these modified sequences of 18–31 nt that can be called as “clean reads” were counted and used for further analysis. Reads were mapped onto the reference genome with default settings and used to search the ncRNA data from Rfam. Annotated expression profiles included miRNAs (based on miRBase, version 21), ribosomal RNAs (rRNAs), small nuclear RNAs (snRNAs), small nucleolar RNAs (snoRNAs), transfer RNAs (tRNAs) (based on Rfam11.0, http://rfam.sanger.ac.uk/; UCSC, http://gtrnadb.ucsc.edu), and Piwi-interacting RNAs (piRNAs; based on piRNABank, http://pirnabank.ibab.ac.in/). Clean reads that can be matched with these databases were screened out for further analysis. On the contrary, the unannotated clean reads were used to predict novel miRNAs in the Mireap software (http://sourceforge.net/projects/mireap/).
miRNA reverse transcription and quantification
We selected two miRNAs for further validation by real-time quantitative reverse transcription PCR (qRT-PCR), according to the expression detected by miRNA sequencing. Total RNA (10 ng) was reverse transcribed using the TaqMan MicroRNA Reverse Transcription Kit (Life Technologies) according to the manufacturer’s instructions with specific primers. The miRNA expression levels were measured using SYBR Green real-time PCR (Life Technologies) following the manufacturer’s instructions. The relative expression ratio of miRNA was normalized by U6, a commonly applied internal control, using 2−ΔCt method. Primers used were as follows: U6 reverse primer (AACGCTTCACGAATTTGCGT), U6 forward primer (CTCGCTTCGGCAGCACA), and unified reverse primer (URP) (TGGTGTCGTGGAGTCG) were from Sangon Biotech (Shanghai, China); has-miR-100-5p and has-miR-148a-3p hairpin primer kits were from GenePharma (Shanghai, China).
Statistical analysis and diagram software
Statistical evaluation and p value calculation of known miRNA differential expression were based on edger analysis with log2 ratio of known miRNA expression of two samples. The results are presented as a scatter plot and mean ± standard error of the mean (SEM). Statistical evaluation of continuous variables was performed using GraphPad Prism, version 6.0. (http://www.graphpad.com/scientific-software/prism/) The p value (<0.05 or <0.01) calculated by two-tailed Student’s t test was considered significant or highly significant. OriginPro software (http://originlab.com/) was also used in drawing diagrams.
Results
Isolation and characterization of exosomes from GC cell lines
After cells were grown for 48 h, exosomes from cell culture–conditioned medium were isolated and purified using ultracentrifugation or commercial reagents. The exosomes had a characteristic vesicle or particle-like shape of nanoscale dimensions. Their diameters ranged from 40 to 100 nm as assessed by TEM (Figure 1(a)). Two of the exosomal membrane proteins on the capsular membranes, CD63 and CD9, were confirmed by Western blot analysis (Figure 1(b)). We also evaluated protein and RNA content in exosomes using BCA and the NanoDrop method, respectively. The yield of exosomal proteins from various GC cells was approximately four-fold higher than that from GES-1 cells (Figure 1(c)). Approximately, 5 mg of proteins was obtained from 30 mL of cell culture medium from GC cell lines, whereas GES-1 cell lines resulted in only 1–2 mg of exosomal protein from 30 mL of cell culture medium. Exosomal RNA was isolated from exosomes derived from 30 mL of cell culture medium of GES-1 and GC cells. The total RNA amounts of MKN45, SGC7901, NCI-N87, AGS, and GES-1 were 596.25, 561.34, 591.26, 684.03, and 209.80 ng, respectively. We observed a three-fold increase in the RNA yield from exosomes isolated from GC cells compared with that of GES-1 cells (Figure 1(d)).

Characterization of exosomes from gastric cancer cell lines. (a) Exosomes isolated from the culture supernatant of cell lines were resuspended in PBS for electron microscopy analyses. Transmission electron microscopy showed round vesicular membrane structures of approximately 100 nm in diameter (scale bar = 100 nm). (b) The expression of exosomal markers CD63 and CD9 was confirmed by Western blotting. The amounts of total protein and RNA contained in different exosomes are shown in (c) and (d), respectively.
Overview of small RNA sequencing data
After exosomal RNA extraction, the next experimental process, including RNA sequence and bioinformatics analysis, is shown in the workflow chart in Figure 2(a). As the quantity of RNA extracted from exosomes was very less, we replicated the cell culture collection of different cell lines and performed sequential isolation of corresponding exosomes. After sufficient RNA was extracted from each cell line, the total RNA was used to build a small RNA library. Overall, we produced a total of five sequencing libraries, representing exosomal RNA from four human GC cell lines (MKN45, SGC7901, NCI-N87, and AGS) and one relatively normal cell line (GES-1). At first, we obtained raw reads, including low-quality reads, contaminants, adaptors, and sequence reads (length < 15 nt). After removing these “noise” sequence reads, we finally obtained clean reads. From the five libraries, we obtained a total of 108,692,021 raw reads, an average of approximately 21.7 million reads per library. And we obtained a total of 81,821,980 clean reads that were considered for further analysis (Table S1). First, the clean reads were mapped on a human reference genome, and the percentage of mappable reads ranged from 55% to 89% (Table S2). After mapping on the human reference genome, we analyzed the distribution of different functional components in the mapping reads by annotating with exons, introns, splicing sites, intergenic regions, upstream/downstream regions, or others based on multiple databases using ANNOVAR software (http://annoar.openbioinformatics.org/en/latest/). This annotation of potential functional components can provide clues to the corresponding sequence, to a certain extent. The statistical results are shown in Table S3. In addition, repeat sequences were also annotated. RepeatMasker (http://repeatmasker.org/) is a program that is widely used in genome analysis and can identify, categorize, and shield repetitive elements, including low-complexity sequences and interspersed repetitive sequences. We performed a Basic Local Alignment Search Tool (BLAST) search of small RNA (clean data) using the RepeatMasker database and identified relevant small RNA repeat sequences. “Repeat” reads were also detected in exosomes. The results are shown in Table S4.
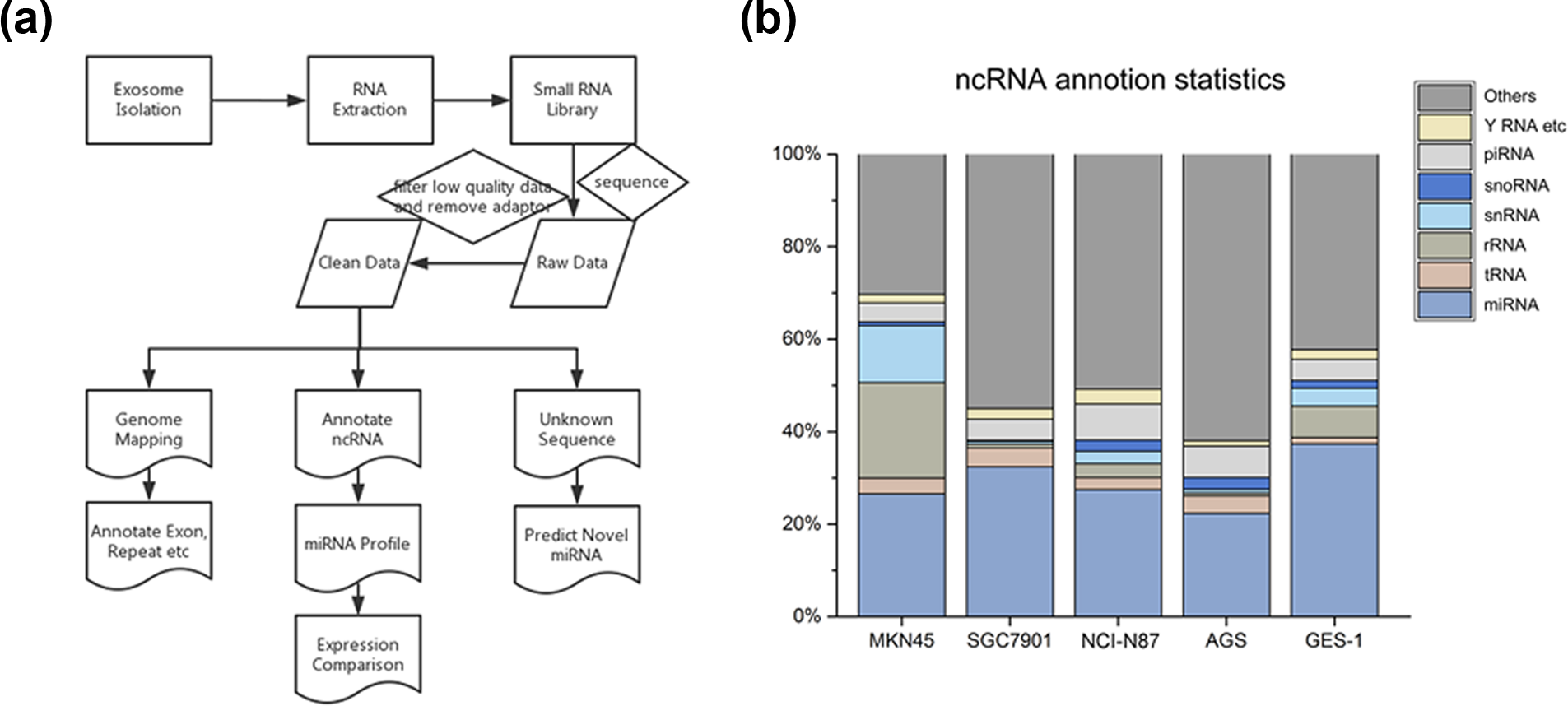
Overview of sequence and bioinformatics analysis and exhibition of non-coding RNA data. (a) Flowchart of the experiment and bioinformatics analysis. (b) Percentage of non-coding RNAs in the sequencing reads.
Annotation of small RNAs from exosomes
Clean reads were derived from raw reads. They were identified based on databases of publicly available ncRNA annotations, including those of miRNAs, tRNAs, rRNAs, snRNAs, snoRNAs, piRNAs, and other small RNAs. Table 1 shows the read number and Figure 2(b) shows the percentage of different types of ncRNAs. As the length of tRNA or rRNA ranges from 50 nt to 500 nt, tRNA and rRNA are not categorized as small RNA. Our purpose in this study was to capture small ncRNAs, which were just 20–40 nt long. Our library preparation protocols guaranteed this. Therefore, the tRNA and rRNA, which were identified in our data, were not complete RNAs, just fragmental part of tRNA or rRNA. The miRNA was the most common among the known sequences, accounting for nearly a quarter of all mappable counts, followed by rRNA, tRNA, snRNA, snoRNA, piRNA, and Y RNA. The miRNAs were used for further analysis. Nevertheless, in all libraries, “others” or unannotated sequence fragments was the most abundant type of small ncRNAs, accounting for half of the mappable reads. This interesting result indicated that a significant number of unannotated sequence fragments, which were secreted into exosomes by the cells, may have potential regulatory functions. The unannotated sequence fragments were used to predict novel miRNA candidates in subsequent analyses.
Read numbers of annotated ncRNAs in total sRNA.
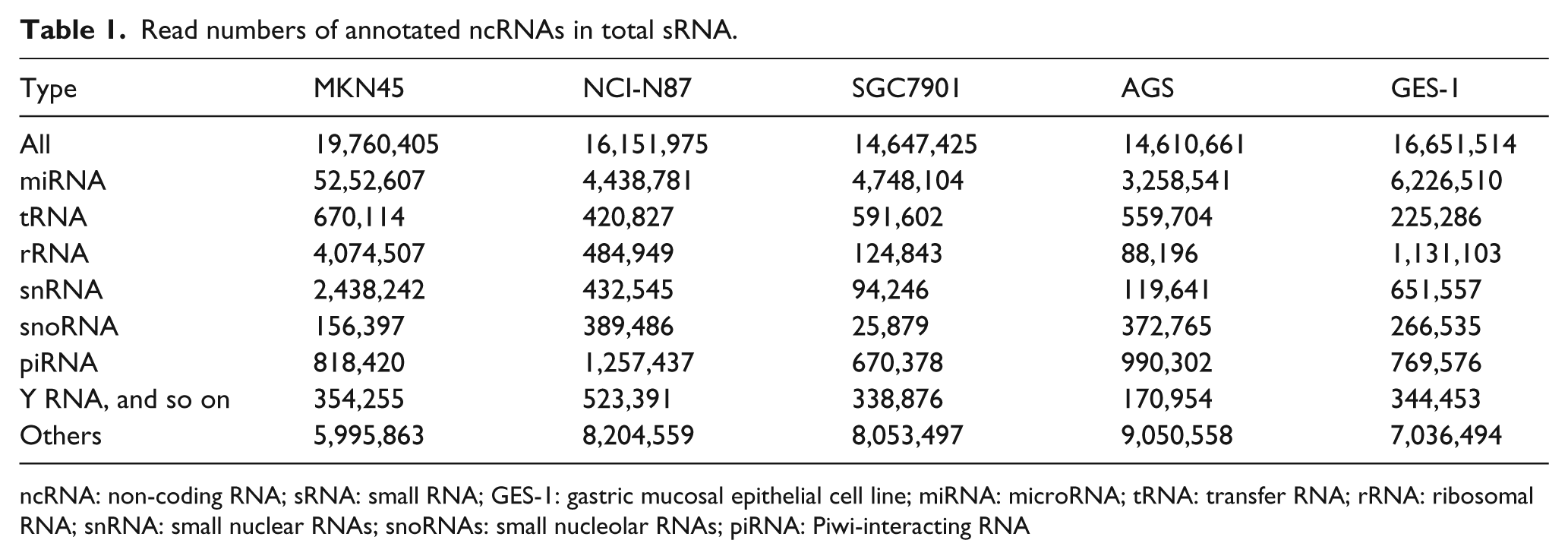
ncRNA: non-coding RNA; sRNA: small RNA; GES-1: gastric mucosal epithelial cell line; miRNA: microRNA; tRNA: transfer RNA; rRNA: ribosomal RNA; snRNA: small nuclear RNAs; snoRNAs: small nucleolar RNAs; piRNA: Piwi-interacting RNA
Known miRNA sequence reads in exosomes
We identified the conserved miRNAs and acquired the information of known miRNAs by comparing our data to miRBase 21, one miRNA database. The clean read count or the absolute sequence read of miRNAs represents the corresponding expression level of each miRNA. Considering the error rate of sequencing, numbers less than 10 reads were removed. The miRNA total reads varied from 10 to 688,126, indicating that the high-throughput sequencing device Illumina HiSeq™ 2500 can detect miRNA fragments regardless of the miRNA expression level. Accordingly, we identified 567, 512, 473, 458, and 306 types of detectable mature miRNAs in exosomes from SGC7901, MKN45, NCI-N87, AGS, and GES-1, respectively. Exosomes derived from GC cells contained significantly more types of miRNAs than exosomes derived from GES-1 cells (Figure 3(a)). Overall, 699 total types of miRNAs were identified, and 266 types were common among GC and GES-1 cells (Figure 3(b)). For comparison, the absolute sequence reads needed to be normalized. The method of miRNA expressed level normalization is RPM—the number of reads per million clean tags. (RPM = number of reads mapped on miRNA/number of reads in clean data × 106). Normalized value is the result of calculation. Normalization showed that exosomes derived from GC cells contained comparatively more miRNA than exosomes derived from GES-1 cells (Figure 3(c)). In each individual library, the 100 most abundant miRNAs made upto 97.38%–97.95% of the detectable miRNA sequences; as a result, the remaining hundreds of low-abundant miRNAs accounted for only 2.05%–2.62%. The five most highly expressed miRNAs, which accounted for almost half of all detectable miRNAs in the five types of exosomes, are shown in Figure 3(d)–(h). In the profile of miRNA expression, hsa-miR-21-5p was the most abundant sequence fragment among all libraries and accounted for 16.09%–34.53% of the total counts of all types of exosomal miRNAs, followed by hsa-miR-30a-5p among all libraries. This result indicates that the distribution of exosome-packed miRNAs was only concentrated in a small part of all miRNAs.

Known miRNA sequence reads in exosomes. (a) The numbers of known miRNAs identified based on publicly available RNA databases in different exosomes. (b) A Venn diagram showing commonly known miRNAs in different exosomes. (c) The normalized values for known miRNAs in different exosomes. (d–h) Expression amount and percentage of each miRNA contained in exosomes. In each diagram, the column chart represents expressional normalized value of each miRNA; the curve chart represents percentage of each miRNA.
Differential miRNA expression in exosomes released from different types of cell lines
The differential expression of miRNA, which is closely associated with the occurrence and development of many diseases, is a hot topic in the field of miRNA research. In this study, we analyzed miRNA profiles of exosomes derived from five types of cell lines and found that a part of miRNA is differently expressed in exosomes from GC cells compared with those in exosomes from GES-1 cells. Normalized values were used to calculate log2 ratios, representing fold change. To visualize miRNA distribution, a scatter diagram was used (Figure 4(a)–(d)). Differentially expressed miRNAs were determined by two criteria: absolute value of log2 (fold change) was greater than or equal to 1 and p value was less than 0.05 based on the edger method. Interestingly, a total of 108 miRNAs were upregulated in the exosomes derived from GC cells compared with GES-1 cells, and four miRNAs, miR-148a-3p, miR-10a-5p, miR-582-3p, and miR-330-3p, were common among the cell types (Figure 4(e)). A total of 23 miRNAs were downregulated, whereas few of them were common among the cell types. Overall, these results show that exosomes derived from cancer cells and normal cells contain a distinct miRNA signature.

Differential miRNA expression profiles of exosomes. (a–d) Scatter diagrams show the distributional discrepancy between two samples, GES-1vsMKN45, GES-1vsSGC7901, GES-1vsNCI-N87, and GES-1vsAGS. (e) There are four common miRNAs among the upregulated miRNAs in the exosomes.
Besides, we also found miRNA differential expression between GC cell lines. Scatter diagrams of differential expression were shown below (Figure S1). Tables S5 and S6 provide a summary of the total number of differences between cell lines, including comparison between GC cell lines and comparison between GES-1 and GC cell lines. The number of differences between the GC cell lines is more than that of the differences between GES-1 and GC cell lines. Considering that tumor cells were established from different patients and different types of tumor, more differences between GC cell lines were reasonable.
However, hsa-miR-148a-3p, one of the common differences we screened out, shows more statistically significant differences in the comparison between GES-1 and GC cell lines than that between cancer cell lines (Table S7).
miRNA validation by qRT-PCR
To validate the findings from deep sequencing, qRT-PCR was performed to detect the significant miRNAs in exosomes released from various cell lines. As stated before, we found four common significantly upregulated miRNAs, including hsa-miR-148a-3p, hsa-miR-10a-5p, hsa-miR-582-3p, and hsa-miR-330-3p. As the expression levels of the latter three miRNAs detected by sequencing were low throughout all libraries, it is difficult to detect them accurately by qRT-PCR (Ct > 35). Therefore, only hsa-miR-148a-3p was used to validate the differences and trends we found earlier by qRT-PCR. In addition, we chose another miRNA of interest, hsa-miR-100-5p, for detection. Hsa-miR-100-5p was more highly expressed than hsa-miR-148a-3p and had at least 1.689-fold change in all GC cells versus GES-1, although it was not a significantly upregulated miRNA. As a result, two miRNAs were selected for further qRT-PCR: hsa-miR-148a-3p and hsa-miR-100-5p. In addition, an internal control, RNU6, was applied in this study. The results generally showed that the expression level of miR-100-5p was higher than that of miR-148a-3p, and levels of both miR-100-5p and miR-148a-3p were significantly upregulated in exosomes derived from most GC cell lines compared with those from GES-1 cells. These results correlate with the sequencing data, suggesting that GC cells can selectively and actively secrete specific miRNAs into the extracellular environment via exosomes (Figure 5).

miRNA validations by qRT-PCR: (a) qRT-PCR results for miR-100-5p expression in exosomes. (b) qRT-PCR results for miR-148a-3p expression in exosomes.
Novel miRNA prediction
In order to identify novel miRNAs in the five libraries, all the clean data were entered into Mireap software (https://sourceforge.net/projects/mireap/), and the predicting process was primarily based on the precursor’s iconic hairpin structure characteristics. Results of preliminary forecasts were saved as .aln format files. We identified a total of 510 novel miRNA candidates: 197 for MKN45 exosome, 86 for SGC7901 exosome, 132 for NCI-N87 exosome, 301 for AGS exosome, and 94 for GES-1 exosome. We screened and optimized candidates for novel miRNAs with greater probability. The following criteria and approaches were used: (1) isomiR was excluded; (2) secondary structure of miRNA sequence was stable, and the free energy of hybridization was less than −20 kcal/mol; (3) checked the reads site in the genome, and novel miRNA should be located in an area between genes; (4) novel miRNAs that met all criteria were marked with asterisks. Accordingly, SGC7901, MKN45, NCI-N87, AGS, and GES-1 contained 31, 80, 64,162, and 45 novel miRNA candidates, respectively. Table 2 shows the top five highly expressed novel miRNAs. The maximal read numbers, representing expression levels of the novel miRNA candidates, ranged from 374 to 3885, which still accounted for a very small proportion of the total reads. Since NGS can detect all fragments of RNA, it is critical to find unknown fragments, especially novel miRNAs. However, the prediction method may not guarantee correct results. The results may be falsely predicted. So, for novel miRNA validation, Northern blot or qPCR methods should be used.
Top five novel miRNA candidates in different exosomes.

miRNA: microRNA; GES-1: gastric mucosal epithelial cell line.
The novel miRNA candidates are located in the intergenic region of the genome.
The novel miRNA candidates are located in the intergenic region of the genome and have a similar miRNA structure.
Discussion
Our studies collected cell-derived exosomes and detected small RNAs in exosomes by NGS. In this study, diverse RNA species were found in exosomes, including miRNA, piRNAs, snoRNAs, and so on. We focused on miRNA, particularly. In this article, has-miR-21 and has-miR-30a were the most abundant in all types of exosomes. They may play potentially critical roles if exosomal target cells take enough of them in. The abundance of the exosomal RNAs from cancer cells or normal cells may be significantly different. We tried to identify them out and recognized two of them, has-miR-148a and has-miR-100. We hope our study can bring other researchers a little enlightenment in exosome studies.
MicroRNA-21 (miR-21) is recognized as an oncomir or oncomiR which is a type of miRNA associated with carcinogenesis, malignant transformation, and metastasis. Dramatic upregulation or significant downregulation of oncomir can be found within cancerous tissues. Our previous research reported that miR-21 was highly expressed in GC tissues and its overexpression could promote cell growth and invasion in GC by targeting phosphatase and tensin homolog (PTEN). 12 Many studies have shown that overexpression of miR-21 is associated with differentiation of tumor, lymph node metastasis, and TNM stage. Moreover, miR-21 detection may be a prognostic method in patients with GC, 13 including miR-21 in serum or plasma.14–16 Cancer cells could secrete more exosomal RNAs, and miR-21 is most abundant in exosomes, which may explain why more miR-21 exists in peripheral circulating blood in cancer patients.
MicroRNA-30a (miR-30a) is generally regarded as a tumor suppressor. Previous studies demonstrated that miR-30 can promote autophagy by beclin 1,17,18 suppress growth and metastasis by targeting metadherin or denticleless protein homolog (DTL),19–21 inhibit epithelial-to-mesenchymal transition by targeting Snai1, 22 and inhibit migration and invasion by downregulating vimentin expression.23,24 Only a few studies reported that miR-30a existed in exosomes, and the levels of miR-30a derived from exosomes in urine, serum or plasma, and saliva could be used as non-invasive biomarkers for screening and diagnosing a certain type of cancer.25–27
Our results indicate that hundreds of particular miRNAs were upregulated in exosomes from cancer cells compared with that from normal cells. Of these differentially expressed miRNAs, microRNA-100 (miR-100) and microRNA-148a (miR-148a) were picked out to validate by qRT-PCR; finally, the trend was similar to that of miRNA sequencing.
In several studies, miR-100 has been described as a tumor suppressor. Downregulation of miR-100 has been detected in breast cancer cells, but stable overexpression of miR-100 strongly reduced oncogene insulin-like growth factor 2 (IGF2) expression and inhibited tumor growth. 28 MiR-100 could induce epithelial-to-mesenchymal transition but inhibit the tumorigenicity, motility, and invasiveness of mammary tumor cells. 29 There is evidence to suggest that miR-100 is required to direct cancer stem-like cell (CSC) self-renewal and differentiation. 30 For endothelial and vascular smooth muscle cells, miR-100 functions as an endogenous repressor by modulating migration, proliferation, tube formation, and sprouting activity. 31 However, its function could be context dependent.
miR-148a is downregulated in several types of tumors, including glioblastoma, 32 hepatocellular carcinoma,33,34 colorectal cancer, 35 and GC. 36 Generally, miR-148a functions as a tumor suppressor in cancer cells.33–36 However, in most studies, miR-148a was detected in specimens of tissues or cell lines. There is limited research involving exosomes containing miR-148a.
We know that mir-21-5p and mir30a-5p are enriched in all kinds of exosomes, while mir-100 and mir-148a are enriched in cancer exosomes. However, we still do not know whether this enrichment is the positive selection or passive process. Moreover, the function of this enrichment phenomenon may be significant and needs further research.
We also detected some sequences of piRNAs and snoRNAs, which also belonged to short, ncRNAs, in exosomes. piRNAs are associated with Piwi protein; snoRNAs are enriched in nucleolus. However, the significant function of exosomal piRNAs and snoRNAs need more research.
Our research made an overview of exosomal RNAs, including both common character and differential expression. We hope that our study can provide other scientists some inspiration about tumor-derived exosome research.
Footnotes
Acknowledgements
B.L. conceived and designed the experiments. J.R., H.L., and Q.Z. performed the experiments and analyzed the data. J.R. and B.L. wrote the article. J.L., L.P., L.S., Q.G., and Z.Z. contributed to the discussion and helped to revise the article. All authors reviewed the article.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
This study was supported by grants from National Natural Science Foundation of China (No. 91529302, No. 81572798, No. 81272749, and 81372231), and Key Projects in the National Science & Technology Pillar Program of China (No. 2014BAI09B03).
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.