
Abstract
Lung cancer is still the leading cause of cancer-related death worldwide, indicating a necessity to develop more effective therapy. Acridine derivatives are potential anticancer agents due to their ability to intercalate DNA as well as inhibit enzymes involved in replication and transcription. Recently, we have evaluated anticancer activity of 32 novel acridine-based compounds. We found that the most effective were tetrahydroacridine and cyclopentaquinoline derivatives with fluorobenzoic acid containing eight and nine carbon atoms in the aliphatic chain. The aim of this study was to determine the molecular mechanisms of compounds-induced cell cycle arrest and apoptosis in human lung adenocarcinoma cells. All compounds activated Ataxia telangiectasia mutated kinase and phosphorylated histone H2A.X at Ser139 indicating DNA damage. Treatment of cells with the compounds increased phosphorylation and accumulation of p53 that regulate cell cycle as well as apoptosis. All compounds induced G0/1 cell cycle arrest by phosphorylation of cyclin-dependent kinase 2 at Tyr15 resulting in attenuation of the kinase activity. In addition, cyclopentaquinoline derivatives induced expression of cyclin-dependent kinase 2 inhibitor, p21; however, tetrahydroacridine derivatives had no significant effect on p21. Moreover, all compounds decreased the mitochondrial membrane potential accompanied by increased expression of Bax and down-regulation of Bcl-2, suggesting activation of the mitochondrial pathway. All compounds also significantly attenuated the migration rates of lung cancer cells. Collectively, our findings suggest a central role of activation of DNA damage signaling in response to new acridine derivatives treatment to induce cell cycle arrest and apoptosis in cancer cells and provide support for their further development as potential drug candidates.
Introduction
Lung cancer is not only the most commonly diagnosed cancers worldwide but is still the leading cause of cancer-related death among both men and women.1,2 The less than 15% of the 5-year survival rate of patients diagnosed with lung cancer indicates an urgent need for searching more effective chemotherapeutic agents.
Acridine derivatives have been explored as potential therapeutic agents for the treatment of cancer due to their non-covalent interaction with DNA by intercalative binding.3,4 Moreover, acridines are able to decrease the activity of enzymes such as topoisomerase I and II that are involved in replication and transcription as well as recombination and chromosome segregation.5,6 The biological activity of acridines is mainly a result of their planarity of aromatic structures, which are able to interact within the DNA structure by intercalation. 7 The act of intercalation induces local structural changes to the DNA, including the unwinding of the double helix and lengthening of the DNA strand, and may cause DNA damage.7,8
Some anticancer agents induce DNA damage, and cellular responses to DNA damage are one of the molecular events associated with cell cycle arrest and apoptosis.9,10 One of the kinases which is involved in the cellular response to DNA double-strand breaks is ATM. 11 It was indicated that this kinase participates in activation of the G1/S cell cycle checkpoint. Moreover, it can forestall damaged cells from going to S phase of cell cycle. 12 During this process, ATM undergoes autophosphorylation at Ser1981 and is recruited to the site of DNA damage to initiate signaling cascade through phosphorylation of multiple proteins. 13 Activated ATM phosphorylates directly histone H2A.X and transcription factor p53 on specific serine. 14 Phosphorylation of H2A.X at Ser139 is considered a hallmark of DNA double-strand breaks. 15 The p53 tumor suppressor protein plays a major role in cellular response to DNA damage. Stability and activity of p53 can be regulated by post-transcriptional modifications such as phosphorylation that affect interaction with the negative regulator MDM2. 16 The activation of p53 can lead to either cell cycle arrest and DNA repair or apoptosis. 17 Some evidences have indicated that p53 binds to single- and double-stranded DNA, recognizes a place of DNA lesion, and promotes ligation of single-stranded DNA. 18
Recently, we have synthesized and evaluated new 16 tetrahydroacridine derivatives and 16 cyclopentaquinoline derivatives with hydrazinonicotinic acid or with fluorobenzoic acid moiety on growth inhibition of human non–small cell lung cancer A549 cells.19–23 Non–small cell lung cancer accounts for more than 80% of all lung cancer cases.1,24 The results of these studies showed that the most effective compounds were tetrahydroacridine and cyclopentaquinoline derivatives coupled with fluorobenzoic acid containing eight and nine carbon atoms in the aliphatic chain. Moreover, the inhibition of cancer cell growth by these compounds was associated with cell cycle arrest in the G0/1 phase and induction caspase 3/7-dependent apoptosis.19,20 However, the precise mechanisms of anticancer activity of these new acridine-based compounds remain unknown. Therefore, the goal of this study was to identify the molecular mechanisms of tetrahydroacridine (compounds 1 and 2) and cyclopentaquinoline (compounds 3 and 4) derivatives with 4-fluorobenzoic moiety involved in inhibition of lung cancer cell proliferation and initiation of cell apoptosis. The chemical structures of compounds evaluated in this study are shown in Figure 1.
Chemical structure of novel tested compounds. Two tetrahydroacridine derivatives (1, 2) and two cyclopentaquinoline derivatives (3, 4) with fluorobenzoic acid moiety are shown. All derivatives have the aliphatic linker comprising eight and nine carbon atoms.
Methods
Cell culture
A549 cells were purchased from the European Collection of Cell Cultures (ECACC). The cells were grown in Dulbecco’s Modified Eagle Medium (DMEM) (EuroClone) supplemented with 10% heat-inactivated fetal bovine serum (FBS; Lonza), 100 U/mL penicillin, and 100 µg/mL streptomycin (EuroClone). Cells were incubated at 37°C in 5% CO2.
Compound treatment
Our new tetrahydroacridine derivatives—compound 1 (IC50 = 15 µM) and compound 2 (IC50 = 10 µM)—and cyclopentaquinoline derivatives—compound 3 (IC50 = 15 µM) and compound 4 (IC50 = 10 µM)—were dissolved in dimethyl sulfoxide (DMSO, Santa Cruz). The final concentration of DMSO in the culture medium did not exceed 0.025%. A549 cells were treated with compound at IC50 concentration and 25 µM. Two concentrations were used for this study based on the previous results evaluating the dose-dependent effects of these compounds on cell cycle arrest and induction of apoptosis.19,20 As a control, the cells were incubated with 0.025% DMSO.
Western blot analysis
A549 cells were treated with tested compounds at indicated concentrations for a period of 0.5, 1, and 24 h. After treatment, the cells were lysed on ice with lysis buffer from a protein isolation kit (Minute Total Protein Extraction Kit Invent Biotechnologies). The protein concentration in cell lysates was determined by Pierce™ BCA Protein Assay Kit. The same amount of protein for each sample was added into 2× Laemmli loading buffer (BioRad) and denatured at 95°C for 5 min. Electrophoretic separation of proteins was performed in 10% polyacrylamide gels (10% acrylamide/bisacrylamide 37.5:1, 1.5 M Tris pH = 8.8; 10% sodium dodecyl sulfate (SDS), 10% ammonium persulfate, tetramethylethylenediamine (TEMED)), and proteins were transferred to a nitrocellulose membrane (BioRad). In order to avoid nonspecific antibody binding, the membrane was blocked in 5% bovine serum albumin (BSA) in Tris-buffered saline (TBS) for 1 h at room temperature. The membranes were immunoblotted with the following primary antibody: phospho-CDK2 (Tyr15) (10A11), phospho-ATM (Ser1981) (D25E5), ATM (D2E2), phospho-p53 (Ser15), p53 (7F5), Bcl-2, Bax (D2E11), β-Actin (D6A8) (Cell Signaling) overnight at 4°C. Next, membranes were washed and incubated with secondary antibody conjugated with horseradish peroxidase (Cell Signaling) for 1 h at room temperature. The immunoblots were developed using ECL Western Blotting Substrate Pico (Pierce™). Reactions were visualized on a photographic film (Primax). Densitometric analysis was performed using GeneTools software (Syngene). Control value was expressed as 1.
H2A.X and p21 immunofluorescence staining
A549 cells were seeded in 6-channel µ-Slide VI0.4 (Ibidi) at density 6000 cells/channel. After 1 h (H2A.X) or 24 h (p21) treatment with tested compounds, cells were fixed in 4% paraformaldehyde and permeabilized in cold methanol for 10 min. After blocking with Image-iT FX Signal Enhancer (Life Technologies) for 30 min, the fixed cells were incubated with rabbit phospho-H2A.X (Ser139) Alexa Fluor 488–conjugated antibody (1:50) or rabbit p21 Alexa Fluor 488–conjugated antibody (1:50) (Cell Signaling) for 1 h. To stain the nuclei, Hoechst 33342 (Invitrogen) was added to the cells and incubated for another 15 min. After washing, the mounting medium (Ibidi) was applied, and cells were photographed with a Nicon Eclipse TE2000S fluorescence microscope using a 40× objective. The total number of nuclei stained with Hoechst and the number of nuclei that expressed green fluorescence from five random microscopic fields per condition were counted. The total number of nuclei for each treatment was expressed as 100%.
Mitochondrial membrane potential
The mitochondrial transmembrane potential was determined using Mito-ID™ Membrane Potential Cytotoxicity assay (Enzo) according to manufacture’s instruction. Briefly, cells were seeded in 96-well plate at density 10,000 cells per well and incubated overnight in standard conditions. After 24 h, cells were treated with tested compounds at IC50 concentrations and higher (25 µM) for 4, 24, and 48 h. Then, 100 µL of Mito-ID MP Dye Loading Solution was added directly into each well. Cells were then incubated for 1 h at room temperature in the dark. The fluorescence of aggregates accumulated in the mitochondria that exhibit orange fluorescence was measured by microplate reader (Synergy H1, Bio-Tek) at excitation 480 nm and emission 590 nm. The results are shown as a ratio of fluorescence of compound-treated cells in relation to the control fluorescence. Fluorescence of control cells is expressed as a 100%. In addition, as a positive control for the dissipation of mitochondrial membrane potential (MMP), cells were incubated with uncoupling mitochondrial agent CCCP (carbonyl cyanide 3-chlorophenylhydrazone) for 30 min at 4 µM. CCCP caused complete loss of MMP in the A549 cells (decrease by 82.3 ± 4.4%, data not shown).
Wound healing assay
A549 cells were seeded on six-well plates at density of 200,000 cells per well. After cultured for 24 h, the confluent cells were wounded by scratching with 100-µL pipette tip. The scratched cells were removed by rising with fresh medium. Next, cells were treated with the tested compounds at IC50 concentrations. Wound closure was monitored using inverted microscope (Opta-Tech) with 4× objective, and photography was done to assess the healing of the wound at time intervals of 0, 24, 48, and 72 h. Images were analyzed by Image J software by monitoring the width of the scratch area at different time intervals to calculate wound closure. The area of wound at 0 time point was express as a 100%.
Statistical analysis
Data are presented as mean ± SD of the number of experiments. The significance difference between control group and compound-treated group was validated by a Student’s paired t-test. A p value below 0.05 (*p < 0.05) was considered statistically significant.
Results
Effect of tetrahydroacridine and cyclopentaquinoline derivatives on ATM activation in A549 cells
ATM is a key regulator of multiple signaling cascades which respond to DNA strand breaks induced by many different factors including chemical agents. These responses involve the activation of cell cycle checkpoints, DNA repair, and apoptosis. In response to DNA double-strand breaks, ATM is rapidly autophosphorylated at Ser1981 and recruited to DNA damage site. 25 To evaluate the role of ATM activation in initiation of a signaling cascade leading to inhibition of cancer cell growth, A549 cells were treated with the compounds for 0.5 and 1 h at concentration of IC50 and 25 µM. Western blotting results revealed that each compound was able to induce phosphorylation of ATM at Ser1981 at both tested concentrations as early as 0.5 h after treatment (Figure 2(a)). Quantitative analysis showed a dose-dependent activation of ATM by cyclopentaquinoline derivatives (compounds 3 and 4). For example, we observed approximately 2.5- and 4-fold increase of ATM phosphorylation levels when cells were treated for 1 h with compound 3 at IC50 and 25 µM concentrations, respectively (Figure 2(b)). Similarly, treatment of cells with compound 4 at concentration of 15 and 25 µM for 0.5 h resulted in two- and threefold increase of ATM phosphorylation levels, respectively. We also observed slightly higher levels of ATM phosphorylation at 25 µM as compared to IC50 in response to tetrahydroacridine derivatives treatment. It seems that compound 3 was the most potent in induction of ATM activation. These results demonstrate that all tested acridine derivatives induce early autophosphorylation of ATM kinase suggesting DNA damage in lung cancer cells.

The effect of tetrahydroacridine (compounds 1 and 2) and cyclopentaquinoline (compounds 3 and 4) derivatives on ATM and p53 activation in A549 cells. (a) Representative images of western blot analysis of A549 cells treated with tested compounds at indicated concentrations for 0.5 and 1 h. (b) Bar graphs show changes in phosphorylation of ATM and p53 after treatment with indicated compounds compared to control cells (Ctr). Control value was expressed as 1. Two concentrations of each compound were tested: IC50 (15 or 10 µM) and 25 µM. Data are presented as a mean ± SD. n = 3.
Effect of tetrahydroacridine and cyclopentaquinoline derivatives on H2A.X phosphorylation in A549 cells
Previously, we have shown that new tetrahydroacridine and cyclopentaquinoline derivatives induce G0/1 cell cycle arrest and apoptosis in A549 cells.19,20 DNA damage is one of the molecular events associated with cell cycle arrest and apoptosis. 26 Histone H2A.X, a variant form of histone H2A, is required for checkpoint-mediated cell cycle arrest and DNA repair following double-stranded DNA breaks.27,28 In response to DNA damage, phosphorylation of H2A.X is rapidly mediated by ATM, Ataxia telangiectasia and Rad3-related (ATR), or DNA-dependent protein kinase (DNA-PK) at Ser139. 29 To elucidate whether acridine derivatives induce double-stranded DNA breaks, we assessed changes in the phosphorylation of H2A.X in single cells in response to treatment using immunofluorescence staining. A549 cells were exposed to tested compounds at concentrations of IC50 range and higher (25 µM) for 1 h. In addition, etoposide, known topoisomerase II inhibitor, was used as a positive control.
Figure 3(a) shows the representative images of A549 cells treated with tested compounds, demonstrating that all compounds induced early phosphorylation of H2A.X in a concentration-dependent manner. Quantitative immunofluorescent analysis showed detection of H2A.X phosphorylation in 16.7 ± 5.7% and 33.4 ± 10.1% of cell nuclei, when cancer cells were cultured in the presence of 25 µM of tetrahydroacridine derivatives, compounds 1 and 2, respectively (Figure 3(b)). Similarly, treatment of cells with cyclopentaquinoline derivatives, compounds 3 and 4, at 25 µM concentration resulted in increased phosphorylation levels of H2A.X in 54.2 ± 10.2% and 26.0 ± 9.0% of cells, respectively (Figure 3(b)). We did not observe detectible fluorescence in nuclei of control cells. For comparison, etoposide treatment of A549 cells induced DNA damage approximately in 50% of cells.

Induction of H2A.X phosphorylation in A549 cells by tested compounds. A549 cells were treated with indicated concentrations of tetrahydroacridine (compounds 1, 2) and cyclopentaquinoline (compounds 3, 4) derivatives for 1 h. Etoposide was used as a positive control. (a) Phosphorylation of H2A.X was examined by immunofluorescent staining using phospho-histone H2A.X (Ser139) antibody conjugated with Alexa Fluor 488. Cell nuclei were stained with Hoechst. Bottom panel shows cell morphology using phase contrast microscopy. 400× magnification. Scale bar indicates 50 µm. (b) Percentage of H2A.X positive cells. The results are expressed as mean ± SD of three independent experiments.
These results demonstrate that new tetrahydroacridine and cyclopentaquinoline derivatives induce early DNA damage in lung cancer cells, thereby causing cell cycle arrest and apoptosis. Moreover, our results revealed that compound 3 was the most potent in induction of H2A.X activation which correlated with the greatest induction of ATM activation by this compound.
Effect of tetrahydroacridine and cyclopentaquinoline derivatives on p53 activation in A549 cells
p53 is an important tumor suppressor protein that plays a major role in cellular response to DNA damage. Upon DNA damage, p53 is phosphorylated at Ser15 by ATM. p53 acts as a nuclear transcription factor and transactivates multiple genes involved in apoptosis and cell cycle regulation. 30
To evaluate the effect of new compounds on p53 activation, A549 cells were cultured in the presence of acridine derivatives for 0.5 and 1 h at IC50 and 25 µM concentration. Western blotting results showed a significantly increased p53 phosphorylation at Ser15 in response to all compounds treatment in dose-dependent manner (Figure 2). For example, we observed three- and fourfold increase in p53 phosphorylation levels in cells treated with compound 3 at 15 and 25 µM, respectively (Figure 2(b)). It seems that cyclopentaquinoline derivatives (compounds 3 and 4) were more effective in induction of p53 activation compared to tetrahydroacridine analogs. These results demonstrate the activation of p53 as downstream signaling molecules in response to DNA damage induced by new acridine derivatives.
Effect of tetrahydroacridine and cyclopentaquinoline derivatives on p21 expression in A549 cells
The tumor suppressor protein p21 is a key component in the induction of cell cycle arrest and apoptosis.31,32 Moreover, p21 is the major transcriptional target of p53 after DNA damage. 33 In association with cyclin-dependent kinase 2 (CDK2) complexes, p21 serves to inhibit kinase activity and blocks progression through G1 to S phase. 34 To evaluate whether new compounds induce cell cycle arrest at G0/1 phase by up-regulating the expression of p21, we determined the expression levels of p21 in single cells by immunofluorescent staining after 24 h treatment at IC50 concentrations. As shown in Figure 4(a), the immunofluorescent images exhibited increased expression of p21 in A549 cells exposed to cyclopentaquinoline derivatives (compounds 3 and 4). Quantitative analysis showed 24.4 ± 5.9% and 27.3 ± 5.4% of nuclei expressing p21 in response to compound 3 and 4 treatment, respectively (Figure 4(b)). However, we did not observe a significant increase in the expression of p21 when A549 cells were cultured in the presence of tetrahydroacridine derivatives (compounds 1 and 2) (Figure 4(a) and (b)). For comparison, etoposide, a DNA-damaging agent, increased the expression of p21 by 65.0 ± 6.3% in A549 cells. Surprisingly, our results revealed different effects of tested tetrahydroacridine and cyclopentaquinoline derivatives on induction of p21 expression. The results suggested that cyclopentaquinoline derivatives induced G0/1 cell cycle arrest by a p21-dependent pathway. In contrast, exposure of cancer cells to tetrahydroacridine derivatives showed no significant effects on p21 expression.

Induction of p21 expression in A549 cells treated with tested compounds. A549 cells were treated with tetrahydroacridine (compounds 1 and 2) and cyclopentaquinoline derivatives (compounds 3 and 4) at IC50 concentrations for 24 h. Etoposide was used as a positive control. (a) Expression of p21 was visualized by immunofluorescent staining using p21 antibody conjugated with Alexa Fluor 488. Cell nuclei were stained with Hochest. Bottom panel shows cell morphology using phase contrast microscopy. 400× magnification. Scale bar indicates 50 µm. (b) Percentage of p21 positive nuclei. Results are expressed as mean ± SD of three independent experiments.
Effect of tetrahydroacridine and cyclopentaquinoline derivatives on CDK2 activation in A549 cells
CDK2 is an important component of the cell cycle machinery. CDK2 activity is regulated by phosphorylation state as well as association with a cyclin subunit and a CDK inhibitor. Inhibitory phosphorylation occurs on Thr14 and Tyr15. 35 The activation of CDK2 complexes requires dephosphorylation of Thr14 and Tyr15 by cdc25 phosphatase. 36 To examine the effect of new compounds on CDK2 activity, we assessed CDK2 phosphorylation at Tyr15. Western blotting results demonstrated a marked induction of CDK2 phosphorylation at Tyr15 after treatment with all compounds. For example, the most effective were compounds 2 and 3 that showed six- to eightfold increased CDK2 phosphorylation compared to control cells (Figure 5). Those results suggest that inhibition of lung cancer cell proliferation by new cyclopentaquinoline and tetrahydroacridine derivatives can be attributed to their ability to attenuate CDK2 activity as a result of enhancement of its phosphorylation at Tyr15.

The effect of tetrahydroacridine (compounds 1 and 2) and cyclopentaquinoline (compounds 3 and 4) derivatives on phosphorylation of CDK2 and expression of Bcl-2, Bax, and p53 in A549 cells. (a) Representative western blotting analysis of A549 cells treated with tested compounds at indicated concentrations for 24 h. (b) Bar graphs show changes in protein activation or expression after treatment with indicated compounds compared to control cells (Ctr). Control value was expressed as 1. Two concentrations of each compound were tested: IC50 (15 or 10 µM) and 25 µM. Data are presented as a mean ± SD. n = 3.
Effect of tetrahydroacridine and cyclopentaquinoline derivatives on Bax and Bcl-2 expression in A549 cells
Apoptosis is regulated by Bcl-2 family proteins that can be subdivided into pro- and anti-apoptotic subsets. Pro-apoptotic protein Bax serves as the ultimate effector of apoptosis by oligomerization to form pores in the mitochondrial outer membrane. In contrast, anti-apoptotic protein Bcl-2 inhibits the initiating steps of apoptosis by binding to BH3 motifs of Bax and blocks its effect. 37 Thus, increase in the levels of Bax and/or decrease in Bcl-2 is a key event in the induction of apoptosis. 38 Therefore, we evaluated the effect of tetrahydroacridine and cyclopentaquinoline derivatives on Bcl-2 and Bax expression in A549 cells. Treatment of cancer cells with all compounds for 24 h significantly decreased the levels of Bcl-2 expression by approximately 60%–80% (Figure 5). In addition, all tested compounds increased the expression of pro-apoptotic Bax by two- to threefold (Figure 5). These results demonstrated increased expression of pro-apoptotic protein Bax and down-regulation of anti-apoptotic protein Bcl-2 in response to treatment of all compounds, suggesting that the induction of apoptosis by new tetrahydroacridine and cyclopentaquinoline derivatives was associated with the activation of the mitochondrial pathway.
Effect of tetrahydroacridine and cyclopentaquinoline derivatives on MMP
Mitochondria play an important role in the apoptotic process. Apoptosis proceeding through the mitochondrial pathway generally displays the permeabilization of mitochondrial membrane, along with a collapse of MMP which is mainly associated with apoptosis. 39
To evaluate the role of mitochondria in compound-induced cell apoptosis, we examined whether tetrahydroacridine and cyclopentaquinoline derivatives cause a loss of MMP. The Mito-ID Membrane Potential Cytotoxicity assay uses a cationic dye that accumulates throughout the cell’s cytosol as a monomer, emitting primarily green fluorescence; with increasing mitochondrial concentration, the dye forms J-aggregates that exhibit orange fluorescence. With mitochondrial damage or loss of membrane potential, the dye cannot accumulate in the mitochondria, which is indicated by decreased or a lack of orange fluorescence. Figure 6 shows kinetics of the changes in MMP in response to tetrahydroacridine (compounds 1, 2) and cyclopentaquinoline (compounds 3, 4) derivatives treatment after 4, 24 and 48 h exposure. All compounds at both tested concentrations (IC50 concentrations and 25 µM) significantly decreased the MMP after 4 h exposure. Treatment of A549 cells with IC50 concentrations resulted in transient decrease of MMP at 4 h and returned to the levels of control range by 24 h. For example, the cells treated with compounds 2 and 4 for 4 h exhibited ~60% of MMP as compared to control cells (Figure 6). In contrast, treatment of cells with 25 µM of compounds caused more profound reduction of MMP that was sustained at all points tested (Figure 6). Compound 2 was the most potent to diminish MMP in A549 cells during the 48 h culture. Exposure of cells to compound 2 at 25 µM resulted in a marked decline in the MMP at 4, 24 and 48 h treatment by 77.5 ± 7.8%, 80.2 ± 7.2%, and 68.2 ± 9.2%, respectively (Figure 6). Data demonstrate that compound-induced changes in MMP were dependent on both the concentrations and the length of cell treatment. The results indicate that the tested compounds affect the mitochondrial function by significantly decreasing the MMP values and causing the depolarization of the mitochondrial membrane. This suggests that the induction of apoptosis by new tetrahydroacridine and cyclopentaquinoline derivatives is associated with the mitochondrial pathway.
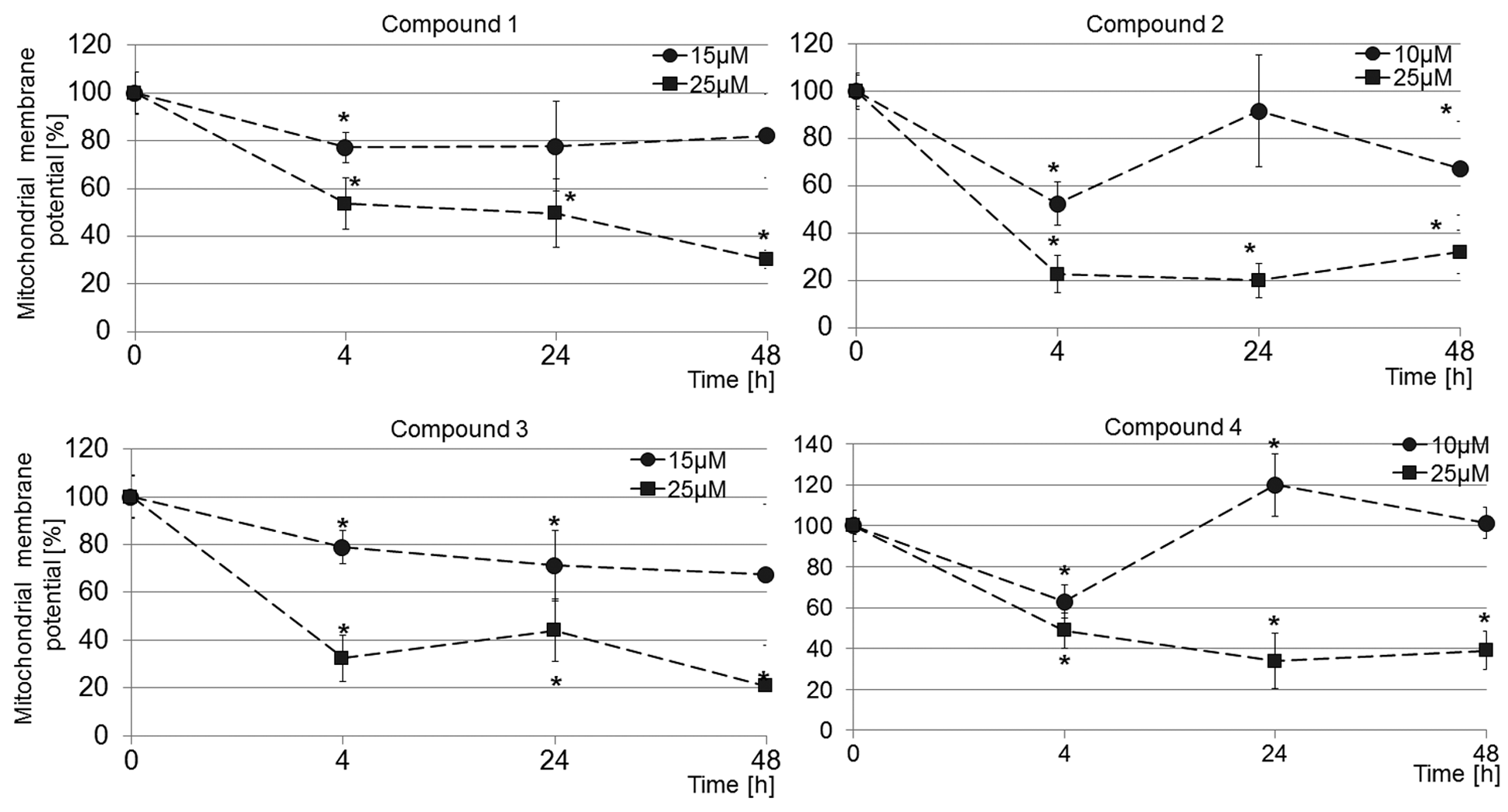
Decrease of mitochondrial membrane potential by tested compounds. The mitochondrial membrane potential was determined using Mito-ID Membrane Potential Cytotoxicity Kit after 4, 24 and 48 h treatment of A549 cells with tetrahydroacridine (compounds 1 and 2) and cyclopentaquinoline derivatives (compounds 3 and 4) at indicated concentrations. Data are expressed as mean ± SD of two independent experiments performed in triplicates.
Effect of tetrahydroacridine and cyclopentaquinoline derivatives on migration of A549 cells
The increased ability of cancer cells to migration plays an important role in tumor invasive and metastasis. The ability of tetrahydroacridine and cyclopentaquinoline derivatives to reduce cell migration was investigated using a classic in vitro wound healing assay. After making a wound, cells were exposed to IC50 concentrations of each compound, the wound was inspected microscopically over time, and the cells that migrated to fill the damage area were photographed at 24, 48, and 72 h. Figure 7(a) shows representative images of wound closure at times 0, 24, 48, and 72 h after wound initiation for both control and compound-treated A549 cells. Wound healing assay showed that all derivatives significantly inhibited cancer cell migration. We observed about 63.7%–73.9% of wound closure in control cells during 72 h (Figure 7(b)). However, when A549 cells were cultured in the presence of compounds 1–4, we observed only 22.1 ± 5.6%, 30.0 ± 13.0%, 18.6 ± 4.7%, and 28.6 ± 12.8% wound closure after 72 h healing, respectively (Figure 7(b)). The results demonstrate a marked attenuation of the migration rates of cancer cells after exposure to new tetrahydroacridine and cyclopentaquinoline derivatives.

Inhibition of cell migration in the presence of tested compounds. (a) Cell migration was evaluated by wound healing assay. Confluents of A549 cells were wounded and treated with or without compounds at IC50 concentrations for 72 h. Cells were photographed using an inverted microscope at the indicated times (t = 0, 24, 48, 72 h). 40× magnification. (b) Graphs represent percentage of wound closure in control and compound-treated cells. Initial area of each wound (t = 0) was expressed as 100%. Data are presented as mean ± SD from four independent experiments.
Discussion
Lung cancer is not only the most commonly diagnosed cancers worldwide but is still the leading cause of cancer-related death.1,2 It is an aggressive tumor with a 5-year survival rate of less than 15%, demonstrating that current therapy is still ineffective.1,2 Therefore, the search for better chemotherapeutic agents with advanced activity against lung cancer is needed.
Acridine derivatives interact with DNA and have ability to inhibit enzymes involved in replication and transcription-such as topoisomerases. 3 The act of intercalation induces local structural changes to the DNA causing the inhibition of replication, transcription, and also DNA repair which subsequently can lead to cell cycle arrest or apoptosis. 40 To explore novel acridine-based compounds as potential chemotherapeutics for treatment of lung cancers, we have recently reported the effects of series of 16 new tetrahydroaminoacridine and 16 cyclopentaquinoline derivatives containing 6-hydrazinonicotinic acid or 4-fluorobenzoic acid moiety on inhibition of human lung cancer cell growth.19,20 Our previous studies have shown that new tetrahydroacridine and cyclopentaquinoline derivatives containing 4-fluorobenzoic acid were much more effective in inhibition of lung cancer cell growth in comparison with compounds with 6-hydrazinonicotinic acid moiety, and their efficacy was correlated with increasing number of carbon atoms in the aliphatic chain. These results revealed that the most effective compounds were tetrahydroacridine and cyclopentaquinoline derivatives with 4-fluorobenzoic acid moiety containing eight and nine carbon atoms in aliphatic chain.19,20 Moreover, the inhibition of cancer cell growth by these compounds was associated with cell cycle arrest in the G0/1 phase and induction caspase 3/7-dependent apoptosis.19,20
However, the precise mechanisms of anticancer activity of these new acridine-based compounds remain unknown. Therefore, the goal of this study was to identify the molecular mechanisms of tetrahydroacridine (compounds 1 and 2) and cyclopentaquinoline (compounds 3 and 4) derivatives with 4-fluorobenzoic moiety which triggered the cell cycle arrest and apoptosis in human lung cancer cells.
The biological activity of acridines is mainly attributed to the planarity of aromatic structures, which can interact within the DNA structure by intercalation and cause DNA damage. 7 Induction of DNA damage signaling is initiated by autophosphorylation of ATM kinase. Activated ATM phosphorylates histone H2A.X and transcription factor p53 on specific serine. 14 The p53 tumor suppressor protein plays a major role in cellular response to DNA damage, and the activation of p53 can lead to either cell cycle arrest and DNA repair or apoptosis. 17 Activated ATM and ATR phosphorylate p53 at Ser15, resulting in the stabilization of p53 and subsequently amplifying the downstream p53 cascade, which modulates both cell cycle and apoptosis. 41 To determine whether the compounds activate genomic DNA damage signaling, lung cancer cells were treated with compounds at IC50 concentration and higher (25 µM). Western blotting analysis was used to detect the markers of DNA damage signaling with specific antibodies recognizing phosphorylated ATM at Ser1981 and phosphorylated p53 at Ser15. The activation of histone H2A.X as a marker of double-stranded DNA breaks was measured in single cells by immunofluorescence staining to detect phospho-H2A.X (Ser139). The results showed that new tetrahydroacridine and cyclopentaquinoline derivatives induced rapid activation of ATM kinase as indicated by autophosphorylation at Ser1981. Moreover, immunofluorescence staining showed that all tested compounds induced early phosphorylation of H2A.X in a concentration-dependent manner indicating double-stranded DNA breaks. Phosphorylated H2A.X serves as a platform for DNA repair enzymes and chromatin-remodeling complexes, and it plays a critical role in the cellular response to double-stranded DNA breaks that involve the activation of cell cycle checkpoints, DNA repair, and apoptosis. 25 In addition, treatment of cancer cells with these compounds induced early phosphorylation of p53 and increased its expression.
Mitochondria play critical roles in cell proliferation and apoptotic processes. The collapse of the MMP is considered to be an indicator of mitochondrial dysfunction and is mainly associated with apoptosis. 42 Intracellular stress signals such as DNA damage may lead to changes in mitochondrial outer membrane potential, and the consequent release of cytochrome c and other pro-apoptotic proteins propagates the apoptotic signal. 43 The studies on the molecular mechanisms of apoptosis showed that the tested compounds affected the mitochondrial function by significantly decreasing the MMP values. Treatment of lung cancer cells with tested compounds at IC50 concentrations resulted in transient reduction of mitochondrial potential after 4 h. However, exposure of cells to 25 µM of compounds caused more profound reduction of MMP that was sustained up to 48 h. The release of cytochrome c to the cytosol is controlled by the members of Bcl-2 family, of which Bax and Bcl-2 have been identified as major regulators. 44 In response to a variety of stimuli including anticancer drugs, pro-apoptotic protein Bax translocates from cytosol to the mitochondria and inserts into the outer mitochondrial membrane, where it allows the release of cytochrome c. In contrast, Bcl-2 blocks cytochrome c efflux by forming a heterodimer with Bax and inhibits its pro-apoptotic effects. 44 Thus, increase in the levels of Bax or decrease in Bcl-2 is a key event in the induction of apoptosis. 38 Our results just showed that new acridine compounds activate p53 which can activate the transcription of a variety of apoptosis-associated genes including Bax when DNA damage exceeds the repair capacity of the cell to induce apoptosis. 30 Analysis of expression of these proteins in cancer cells treated with tested compounds demonstrated increased expression of pro-apoptotic protein Bax and down-regulation of anti-apoptotic protein Bcl-2. Our results also showed that changes in the levels of Bax and Bcl-2 expression were associated with the activation of p53 in A549 cells in response to treatment with all tested compounds. These results suggested that the induction of apoptosis by new tetrahydroacridine and cyclopentaquinoline derivatives coupled with 4-fluorobenzoic acid was due to the activation of the mitochondrial pathway. Performed analysis showed that all tested compounds can be considered as potent apoptosis inducers because they enhance pro-apoptotic signal via Bax induction while decreasing anti-apoptotic effect of Bcl-2. It has been reported that quinacrine (9-aminoacridine derivative) inhibits cell proliferation and induces apoptosis of breast cancer cells as a result of increased activation of p53 and expression of Bax and p21. 45 Previously, we have demonstrated that new tetrahydroacridine and cyclopentaquinoline derivatives with 4-fluorobenzoic acid inhibit cell proliferation by cell cycle arrest in the G0/1 phase.19,20 The cell cycle progression is controlled by cyclin and cyclin-dependent kinase (CDK) which promotes the cell entering into the cell cycle, whereas CDK inhibitor (CKI) functions to arrest the cells.46,47 Progression from the G1 phase into the S phase is controlled by CDK2 and CKI, p21 Waf1/Cip1, and p27 Kip1. 48 CDK2/cyclin E kinase activity is important for the G1 to S transition and phosphorylation of the Rb protein. CDK2 activity is also regulated by phosphorylation state as well. The activation of CDK2 complexes requires dephosphorylation of Thr14 and Tyr15 by cdc25 phosphatase and phosphorylation of Thr160. 36 Phosphorylation of CDK2 at Tyr15 leads to inhibition of the kinase activity.36,49
To determine the mechanisms of compound-induced cell cycle arrest in G0/1 phase, we examined the effect of tested compounds on CDK2 activity. Western blot analysis of A549 cells treated with compounds for 24 h revealed that exposure of cancer cells to new acridine derivatives markedly increased the phosphorylation of CDK2 at Tyr15 resulting in inhibition of the kinase activity and subsequent inhibition of cell progression from G1 to S phase.
Inhibition of CDK2-cyclin complexes can also be attributed to association with CKI, p21. 48 Therefore, we have evaluated the effects of the compounds on the expression levels of p21 in single cells using immunofluorescence staining. Treatment of cancer cells with cyclopentaquinoline derivatives (compounds 3 and 4) showed a significant increase in the expression of p21 in the nuclei. In contrast, exposure of cancer cells to tetrahydroacridine derivatives (compounds 1 and 2) had no significant effects on p21 expression levels. The study on the molecular mechanisms of inhibition of cancer cell proliferation demonstrated that tested tetrahydroacridine and cyclopentaquinoline derivatives induced G0/1 cell cycle arrest by inhibition of CDK2 activity due to increased phosphorylation at Tyr15. In addition, cyclopentaquinoline derivatives induced G0/1 cell cycle arrest by increasing the expression of CKI, p21, in contrast to tetrahydroacridine derivatives. Transcription of p21 can be induced by the activation of p53 in response to DNA damage. 43 Surprisingly, our data showed that cyclopentaquinoline as well as tetrahydroacridine derivatives activated p53 in cancer cells; however, only cyclopentaquinoline derivatives inhibited cell proliferation by p21-dependent pathway.
The cell migration plays an important role in tumor invasiveness and metastasis. Lung cancer cells have highly invasive and metastasis properties with an ability to migrate into surrounding tissues. 50 Therefore, inhibition of cancer cell migration, in addition to inhibition of cell proliferation and induction of apoptosis, is a very important aspect of anticancer activity of potential drugs. The cell migration was measured by wound healing assay during 72 h. The results demonstrated a marked attenuation of the migration rates of cancer cells in the presence of tetrahydroacridine derivatives as well as cyclopentaquinoline derivatives. It is important that in addition to inhibition of cell proliferation and induction of apoptosis, these compounds also were very effective in attenuation of cancer cell migration. This observation is consistent with other studies demonstrating that quinacrine (9-aminoacridine derivative) reduced migration of breast cancer cells. 45
In conclusion, the results indicate that novel tetrahydroacridine and cyclopentaquinoline derivatives with 4-fluorobenzoic acid induced double-stranded DNA breaks and activated DNA damage signaling by phosphorylation of ATM and HA2.X that triggered p53 activation. Induction of apoptosis was associated with the activation of mitochondrial pathway as a result of decrease in the MMP as well as down-regulation of Bcl-2 and up-regulation of Bax expression. All compounds induced cell cycle arrest in G0/1 phase by inhibition of CDK2 activity. In addition, cyclopentaquinoline derivatives attenuated cell proliferation by p21-dependent mechanism. These new compounds are great candidates for further evaluation in preclinical studies because they inhibit lung cancer cell growth by induction of apoptosis as well as cell cycle arrest and attenuate cell migration.
Footnotes
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: Financial support by grant 502-03/3-015-01/502-34-040 and 503/3-015-01/503-31-002 from the Medical University of Lodz, Poland, is gratefully acknowledged.