
Abstract
The TP53 gene remains the most frequently altered gene in human cancer, of which variants are associated with cancer risk, therapy resistance, and poor prognosis in several tumor types. To determine the true prognostic value of TP53 variants in non–small cell lung cancer, this study conducted further research, particularly focusing on subtype and tumor stage. Therefore, we determined the TP53 status of 97 non–small cell lung cancer adenocarcinoma patients using next generation deep sequencing technology and defined the prognostic value of frequently occurring single nucleotide polymorphisms and mutations in the TP53 gene. Inactivating TP53 mutations acted as a predictor for both worse overall and progression-free survival in stage II–IV patients and patients treated with DNA-damaging (neo)adjuvant therapy. In stage I tumors, the Pro-allele of the TP53 R72P polymorphism acted as a predictor for worse overall survival. In addition, we detected the rare R213R (rs1800372, minor allele frequency: 0.0054) polymorphism in 7.2% of the patients and are the first to show the significant association with TP53 mutations in non–small cell lung cancer adenocarcinoma patients (p = 0.003). In conclusion, Our findings show an important role for TP53 variants as negative predictors for the outcome of non–small cell lung cancer adenocarcinoma patients, especially for TP53 inactivating mutations in advanced stage tumors treated with DNA-damaging agents, and provide the first evidence of the R213R G-allele as possible risk factor for non–small cell lung cancer.
Introduction
In recent years, remarkable progress has been made in improving treatment modalities of lung cancer, with a shift from more general to targeted treatments depending on the molecular background of individual tumors. However, despite these promising developments, this devastating disease remains the leading cause of cancer-related death worldwide. 1 Approximately 75%–85% of all lung malignancies are currently classified as non–small cell lung cancer (NSCLC), for which the predicted 5-year survival rate for newly diagnosed cases is still below 20%. 1 Currently, prognosis of NSCLC patients is mainly based on specific clinicopathologic factors and tumor stage. Nevertheless, within one subgroup, there are considerable differences in patient outcome (i.e. progression-free survival (PFS) and overall survival (OS)), even when patients receive identical treatments. 2 A more accurate prognostic assessment, incorporating additional molecular biomarkers, is therefore needed.
The TP53 gene remains the most frequently altered gene in human cancer, and both mutations and single nucleotide polymorphisms (SNPs) have been shown to alter p53’s normal functions. P53 ensures regulation of the cellular response to a variety of stress signals by inducing senescence, cell cycle arrest, and/or apoptosis, thereby playing an important role as tumor suppressor protein. Our group recently reviewed the currently available data on the prognostic and predictive value of TP53 genetic alterations in NSCLC, particularly focusing on the TP53 mutational status and the TP53 R72P polymorphism (rs1042522), of which the Pro variant is associated with reduced apoptotic inducing capacities.3,4 Although these data showed a clear influence of TP53 variants on the patients’ prognosis, further research is warranted, particularly focusing on tumor stage. In addition, only limited data of the effect of TP53 variants on PFS is available.
In this study, next generation deep sequencing technology was used to assess genetic alterations in the TP53 gene, resulting in a higher sensitivity than that achieved by conventional Sanger sequencing mostly used in previous studies. 3 In addition, deep sequencing offers the possibility to detect mutations in the presence of normal cell contamination, which often occurs after DNA isolation from formalin-fixed paraffin-embedded (FFPE) tissue by macrodissection techniques. In order to better assess the mutational spectrum of the TP53 gene, the whole coding region consisting of exons 2–11 was sequenced, including splice site regions.
Our first objective was to map all TP53 variants, including SNPs, in a NSCLC adenocarcinoma (AC) patient group and determine possible associations between the SNPs identified in this study, R72P (rs1042522) and R213R (rs1800372), and TP53 mutations. Second, the presence of these variants was linked to clinicopathological characteristics of the NSCLC patients. Third, the prognostic value of frequently occurring SNPs and the TP53 mutational status was determined in both univariate and multivariate OS and PFS analysis.
Material and methods
Patient cohort
The study cohort consisted of 97 patients diagnosed with stage I–IV NSCLC. Patients were selected by an independent researcher based on histological subtype (AC). Tissue samples from tumor resections were obtained from the Antwerp University Hospital Tumorbank, which is funded by the National Cancer Plan, and Onze-Lieve-Vrouw hospital Aalst (Belgium) (see Table S1, Supplemental Data 1, which describes the patients’ characteristics). Patients were diagnosed between 2005 and 2014. EGFR/ALK status was available for only a limited number of patients and therefore not taken into account. Tissue specimens were fixed in 4% formaldehyde for 6–18 h and paraffin embedded on a routine basis. A formal consent was not required for this type of study.
DNA extraction and quantification
DNA was extracted from 5 to 10 FFPE tissue slides (10 µm thick), depending on the size of the tumor region. Tumor regions were determined by an experienced pathologist based on H&E (hematoxylin & eosin)-stained reference slides and enriched by macrodissection. DNA was isolated using the QIAmp DNA FFPE tissue kit (Qiagen, Venlo, the Netherlands) according to the manufacturer’s instructions. DNA concentrations were measured on a Qubit 2.0 fluorometer using the Qubit dsDNA BR assay (Invitrogen, Merelbeke, Belgium). The purity (A260/280 ratio and A260/230 ratio) was measured using the NanoDrop ND-1000 spectrophotometer (ThermoFisher Scientific, Gent, Belgium).
Multiplex amplification of specific targets for resequencing
TP53 sequencing was performed on genomic tumor DNA of 69 samples using the commercially available TP53 multiplex amplification of specific targets for resequencing (MASTR™) with MID for Illumina MiSeq kit (Multiplicom, Niel, Belgium) according to the manufacturer’s instructions. An additional 28 samples were sequenced using the custom-designed TP53-cMET MASTR kit (Multiplicom) according to the assay specific protocol provided by the manufacturer. These samples were obtained from another study in our group, particularly focusing on the role of cMET in NSCLC, and were included in this study to increase sample size and statistical power. A total of 10 NSCLC patient samples and one NSCLC cell line (NCI-H596, TP53G245C), obtained from the American Type Culture Collection (ATCC; Rockville, USA), were run with both MASTR kits as a control and resulted in identical results.
Sequencing was performed on a MiSeq Sequencing system using a MiSeq Reagent Kit v2 (500 cycles) (Illumina, San Diego, USA).
Using VariantDB, 5 an in-house annotation and filtering tool, single nucleotide variants (SNVs) were annotated to the TP53 NM_000546 reference sequence. SNVs present in the dbSNP137 and 1000 Genomes Project databases were defined as SNPs, and SNPs with a minor allele frequency (MAF) lower than 0.05 were considered as rare variants. The reads were visualized using the Integrative Genomics Viewer software (IGV, version 2.3.67), aligned to the human reference genome (hg19, NCBI build 37). Using the MUT-TP53 2.0 tool, 6 TP53 SNVs were presented according to the Human Genome Variant Society (HGVS) recommendations. In addition, frequencies of the variants in the UMD-p53 database are presented, as well as functional information of mutant p53.
Statistical analysis
Associations between the TP53 mutational status, R72P or R213R polymorphisms, and clinical and pathological parameters of NSCLC patients were investigated by χ2 analysis or Fisher exact test (when appropriate) for categorical variables and the Student’s t-test (two groups) or one-way analysis of variance (ANOVA; >2 groups) for continuous variables.
Prognostic value was assessed by survival analysis. PFS was defined as the time until disease progression occurred. OS was defined as the time until cancer-related death occurred. Univariate OS and PFS probability were estimated using the Kaplan–Meier method. Statistical significance was determined using the log-rank test.
A post hoc statistical power analysis was performed to determine whether the study compromised sufficient subjects to detect significance for univariate survival analysis (log-rank test) using MedCalc (α = 0.05 and power ⩾ 0.80).
In addition to univariate analysis, a multivariate Cox proportional hazards model was fitted to identify independent prognostic markers, presented as a hazard ratio (HR) and its 95% confidence interval (CI). All analyses were performed using SPSS version 23, and significance was reached if p < 0.05 (two-tailed).
Results
TP53 mutational pattern
Next generation deep sequencing identified a heterogeneous set of genetic variants in the TP53 gene. Three SNPs were detected: the common R72P polymorphism (rs1042522; MAF(G):0.4571), of which the Pro variant (present in 34.1% of the patients, Table 1) is associated with reduced apoptotic inducing capacities compared to the Arg variant; the rare exonic variant R213R (rs1800372, MAF: 0.0054), present as a heterozygous genotype in 7.2% of the patients (Table 1); and P36P (rs1800370; MAF: 0.0076), which was identified in one patient.
Patient characteristics TP53 variants.

Inactivating mutations: single nucleotide variants (nonsense, missense), deletions.
In addition, 44 SNVs were detected in 41 patients (see Table S2, Supplemental Data 1, which presents each mutation and its activity). These consisted of 39 missense mutations and five nonsense mutations. One patient harbored both a nonsense and a missense mutation (R196X/R110L), and one patient harbored three missense mutations (G245C/G266V/R280G). Finally, we identified one in-frame deletion (p.P177_C182delPHHERC, exon 5), one frameshift deletion (p.L93fs, exon 4), and one insertion (c.1162_1163insT, exon 11). The latter was not taken into account as inactivating mutation. In total, 43 patients (44.3%) harbored at least one inactivating mutation in the TP53 gene (Table 1).
Relationship between clinicopathological parameters, prognosis, and TP53 variants
TP53 variants were not associated with clinicopathological parameters
In order to reach sufficient statistical discriminatory power, we combined the Pro-allele of TP53 codon 72 for further analysis (Arg/Arg vs Arg/Pro|Pro/Pro). Neither the R72P polymorphism nor the R213R polymorphism or the TP53 mutational status were associated with the clinicopathological parameters listed in Table 2 (age, gender, tumor stage, T-classification, differentiation, metastasis, or smoking status).
Association of TP53 polymorphisms and TP53 mutation status with clinicopathological characteristics.

WT: wild-type.
At time of diagnosis
Initially and/or after progression.
R72P was not associated with the presence of TP53 mutations. However, the heterozygous AG-genotype of the R213R polymorphism was significantly associated with TP53 mutations (p = 0.003; Table 2). One patient harbored one missense and one nonsense TP53 mutation in association the R213R polymorphism, while the other patients harbored one missense mutation (see Table S3, Supplemental Data 1, which presents each R213R-associated mutation).
The prognostic value of the R72P Pro-allele
Univariate survival analysis did not show any difference in OS or PFS with respect to the R72P polymorphism (Arg/Arg vs Arg/Pro|Pro/Pro) in stage I–IV patients (Figure 1).

Kaplan–Meier survival curves for OS and PFS according to R72P status: patients were grouped by either Arg/Arg genotype (blue) or Arg/Pro|Pro/Pro genotype (green). Patients were additionally stratified by stage I and stage II–IV status. Significance was reached when p < 0.05 and was determined by log-rank testing (n = 97).
When taking tumor stage into account, univariate analysis showed a trend toward worse OS in stage I NSCLC patients for carriers of the Pro-allele (Arg/Pro or Pro/Pro, p = 0.069), while this effect was not observed in stage II–IV patients. Grouping by tumor stage did not alter the effect on PFS (Figure 1).
TP53 mutational status significantly affects both OS and PFS of NSCLC patients, depending on tumor stage and treatment
Univariate survival analysis showed that the presence of an inactivating TP53 mutation was significantly associated to poor OS in stage I–IV patients (p = 0.004; Figure 2). However, when grouping patients based on tumor stage, the significant effect was no longer observed in stage I tumors (p = 0.793), while stage II–IV tumors were even more significantly associated to worse OS (p = 0.001).

Kaplan–Meier survival curves for OS and PFS according to TP53 mutational status. Patients were grouped by either TP53 wild-type (blue) or TP53 mutant (green). Patients were additionally stratified by stage I and stage II–IV status. Significance was reached when p < 0.05 and was determined by log-rank testing (n = 97).
A possible cause might be the difference in therapeutic strategy between stage I and stage II–IV tumors within our patient group. Treatment data were available for 78/97 patients and showed that only 20.7% of stage I patients were treated with neo-adjuvant and/or adjuvant DNA-damaging agents (platinum-based chemotherapy and/or radiotherapy) in contrast to 83.7% in stage II–IV patients. TP53 status did not affect OS in the non-treated group (p = 0.870), similar to stage I patients. However, mutant TP53 was strongly associated to poor OS in the treated group (p < 0.001), similar to the stage II–IV patients (Figure 3).

Kaplan–Meier survival curves for OS according to TP53 mutational status. Patients were grouped by either TP53 wild-type (blue) or TP53 mutant (green). Patients were stratified by treatment with (neo)adjuvant DNA-damaging agents (platinum-based chemotherapy and/or radiotherapy). Significance was reached when p < 0.05 and was determined by log-rank testing (n = 78).
A significant effect for the presence of an inactivating TP53 mutation on PFS was observed only in stage II–IV patients (p = 0.039), but not in stage I patients (p = 0.874), indicating a more prominent role for the TP53 mutational status in late stage tumors on both OS and PFS. However, these results were underpowered due to sample size.
Independent predictors of OS and PFS
In addition to univariate analysis, we fitted a Cox proportional hazards model with TP53 mutational status and R72P status as predictor for OS and accounting for potential confounders including gender, age, metastasis, N-stage, and tumor stage (Table 3). In addition, we included the R213R status, although univariate analysis did not indicate any prognostic value for this SNP (data not shown).
Independent predictors of OS en PFS (multivariate Cox regression model).
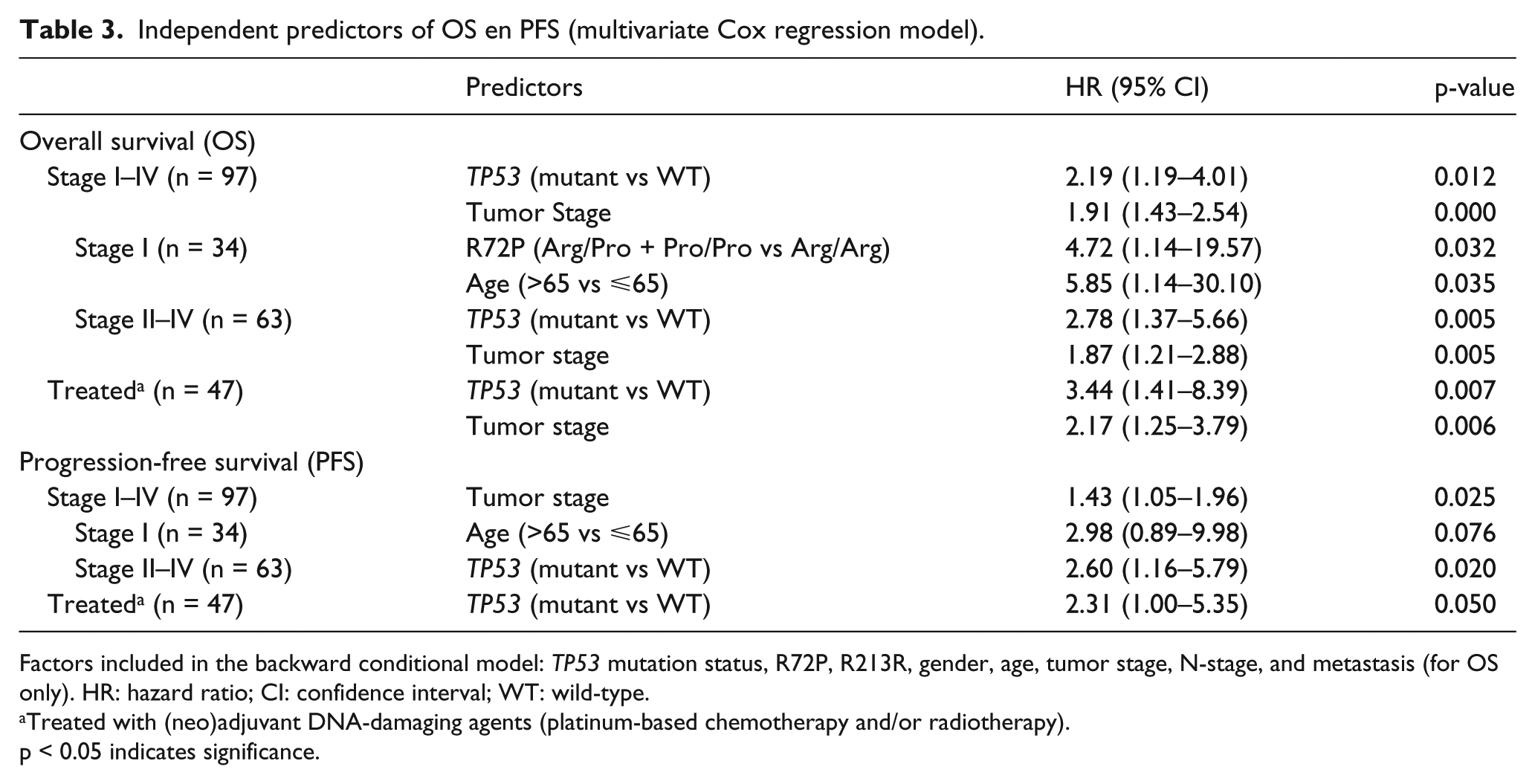
Factors included in the backward conditional model: TP53 mutation status, R72P, R213R, gender, age, tumor stage, N-stage, and metastasis (for OS only). HR: hazard ratio; CI: confidence interval; WT: wild-type.
Treated with (neo)adjuvant DNA-damaging agents (platinum-based chemotherapy and/or radiotherapy).
p < 0.05 indicates significance.
Stepwise backward model building showed that TP53 mutational status had a significant effect on OS in stage I–IV patients with a HR of 2.19 (95% CI: 1.19–4.01; p = 0.012). Tumor stage acted as a significant confounder (HR: 1.91; 95% CI: 1.43–2.54: p = 0.000), which was expected from the univariate analysis, showing a difference between stage I and stage II–IV tumors. The HR for TP53 mutations increased to 2.78 (95% CI: 1.37–5.66; p = 0.005) in stage II–IV tumors, but still had to be accounted for by tumor stage (HR: 1.87; 95% CI: 1.21–2.88; p = 0.005). Due to the limited sample size, further discrimination by tumor stage was not possible. In stage I tumors, no significant prognostic value remained for TP53 mutation status. However, the R72P Pro-allele acted as a strong predictor for worse OS (HR: 4.72; 95% CI: 1.14–19.57; p = 0.032) in stage I tumors, accounting for age.
When patients were stratified by treatment, we observed that TP53 mutation status had an effect on OS in the neo-adjuvant and/or adjuvant treated group with a HR of 3.44 (95% CI: 1.41–8.39; p = 0.007) with tumor stage acting as significant confounder (HR: 2.17; 95% CI: 1.25–3.79; p = 0.006), similar to the effect observed in stage II–IV patients (Table 3). No effect for TP53 variants was observed in the non-treated patient group.
A Cox model was fitted for TP53, R72P, and R213R status as predictors for PFS, accounting for gender, age, N-stage, and tumor stage (Table 3). Stepwise backward modeling showed that tumor stage acted as a significant independent predictor for poor PFS in stage I–IV patients (HR: 1.43; 95% CI: 1.05–1.96; p = 0.025). However, its prognostic value was not retained in stage II–IV patients, for which the TP53 mutation status acted as a significant and independent predictor for PFS (HR: 2.60; 95% CI: 1.16–5.79; p = 0.020). A similar effect was observed in the (neo)adjuvant-treated patients (HR: 2.31; 95% CI: 1.00–5.35; p = 0.050) (Table 3).
No effect on PFS from TP53, R72P, or R213R status was observed in stage I patients, nor the untreated patient group.
Discussion
The prognostic value of TP53 variants in NSCLC has been frequently studied, but has often led to conflicting results, especially for the role of the R72P SNP. Therefore, more in-depth studies are warranted, particularly focusing on NSCLC subtypes and possible differences between tumor stages. 3
In this study, we focused on a Caucasian (Belgian) NSCLC AC patient group consisting of stage I–IV tumors. In accordance with previous studies, inactivating TP53 mutations were detected in 44% of all patients. 3 In addition, three SNPs were detected, including the frequently occurring R72P polymorphism of which the Pro variant is associated with reduced expression of apoptotic-related target genes. 4 We did not observe any association of the R72P SNP with TP53 mutations, although this was previously reported by others. 7 The rare synonymous P36P SNP was detected in one patient, and has previously been reported to affect mouse double minute 2 homolog (MDM2)-mediated control of TP53 messenger RNA (mRNA) translation. 8 Interestingly, we detected the rare R213R (rs1800372) polymorphism in 7.2% of the patients and are the first to show the significant association with TP53 mutations in NSCLC AC patients. Ganci et al. 9 previously reported R213R positive head and neck squamous cell carcinoma (SCC) patients, who were all heterozygous for this locus. In addition, they showed higher expression of the synonymous R213R polymorphic allele compared to the wild-type allele, which was related to TP53 LOH (loss of heterozygosity) positive status, which is frequently observed in the presence of TP53 mutations. 10 However, TP53 mutations were only detected in two out of five R213R positive patients, taking into account that their study was limited to exons 5–8. Since we observed R213R-associated mutations in exon 4, and Ganci et al. detected TP53 LOH in these exon 5–8 TP53 wild-type patients, there is a strong possibility that these patients carry on other mutations on exons different than the ones studied by the authors. Despite the association of R213R with inactivating TP53 mutations, which acted as an independent predictor for worse OS and PFS, R213R did not act as a prognostic marker. In addition, the outcome of the multivariate model was not influenced by the R213R polymorphism. Consequently, R213R shows limited potential as a predictor for patients’ outcome. Nonetheless, R213R could acts as a valuable marker for the assessment of the risk for NSCLC development, possibly due to the increased susceptibility for TP53 mutations and high frequency in our patient group compared to the general population (MAF: 0.0054).
TP53 inactivating mutations were not associated with any of the patients’ clinicopathological data, including invasiveness, lymph node infiltration, or metastasis. This shows that TP53 mutations did not seem to affect tumor aggressiveness which could have accounted for the worse OS and PFS observed in our patient group. The fact that the prognostic role for TP53 mutations was unique to stage II–IV tumors might depend on the difference in therapeutic strategy for stage I and stage II–IV patients. Surgical resection remains the main therapeutic option for stage I patients, and most of the stage I patients in our study cohort did not receive (neo)adjuvant therapy, while advanced stages were mostly treated with DNA-damaging agents. Previous studies have shown that in patients who received platinum-based adjuvant chemotherapy, the presence of mutant type TP53 showed a tendency for shorter survival compared to wild-type TP53, depending on the specific type of mutation.11–13 Hence, the stronger prognostic value we observed for TP53 mutations in advanced stage NSCLC could be due to therapy resistance/failure, supported by the negative prognostic effect of TP53 mutations in the (neo)adjuvant-treated patients of our study population. The currently available data on the prognostic value of TP53 mutations, recently reviewed by Deben et al., 3 indicated the importance of subtype and tumor stage. Previous studies have shown that the prognostic value of TP53 mutations was limited to AC patients, and not observed in SCC patients, consistent with our results in AC.14–16 Both Ahrendt et al. 17 and Chien et al. 7 reported a significantly worse OS in stage I patients harboring a TP53 mutation, but not in stage II–III patients. However, these studies did not take subtype into account, which is clearly an influencing factor as discussed above. This could account for the contradictory results observed in this study, which was limited to AC tumors. Only limited studies were available on the predictive role of TP53 mutations on PFS, reporting no effect of TP53 mutations on PFS. However, these studies failed to categorize by tumor stage or subtype.12,18
The Pro-allele of the R72P polymorphism acted as a strong predictor for poorer OS in stage I tumors in our study cohort. One previous study took tumor stage into account and found a similar worse OS in the presence of at least one Pro-allele in stage I tumors, although this effect was not significant and subtype was not taken into account. 7 The R72P Pro-allele did not affect PFS in our patient cohort, even when grouped by tumor stage. Han et al. 19 and Liu et al. 20 showed a significant association of the Pro-allele with poorer PFS, limited to stage III–IV patients. On the contrary, Shiraishi et al. 21 reported a significant better PFS for patients harboring the Pro/Pro genotype. Again, these studies included both AC and SSC subtypes, making it difficult to compare these findings to our results.
In conclusion, we observed a high frequency of the R213R variant in our patient population, indicating that the R213R G-allele might act as an important risk allele for NSCLC, possibly due to its strong association with TP53 mutations. In addition, our findings clearly show an important role for TP53 variants as predictors for a worse outcome of NSCLC AC patients, especially for TP53 inactivating mutations in advanced stage tumors, possibly due to therapy resistance/failure. Restoring wild-type p53 function using compounds like APR-246 (PRIMA-1MET) or exploiting the presence of mutant p53, as previously discussed, could therefore prove to be promising therapeutic strategies which could greatly improve patients’ outcome.3,22
Footnotes
Acknowledgements
The authors would like to thank Multiplicom for funding the TP53 MASTR™ kit and technical assistance.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: C.D., J.J., and N.V.D.S. were funded by the Agency for Innovation by Science and Technology, Flanders (grant numbers 111063, 120822, 120912). A.W. is funded by Research Foundation Flanders (grant number 1297813N).
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.