
Abstract
Current standard chemotherapy for late stage ovarian cancer is found unsuccessful due to relapse after completing the regimens. After completing platinum-based chemotherapy, 70% of patients develop relapse and resistance. Recent evidence proves ovarian cancer stem cells as the source of resistance. Therefore, treatment strategy to target both cancer stem cells and normal stem cells is essential. In this study, we developed a novel chalcone derivative as novel drug candidate for ovarian cancer treatment. We found that methoxyphenyl chalcone was effective to eliminate ovarian cancer cells when given either as monotherapy or in combination with cisplatin. We found that cell viability of ovarian cancer cells was decreased through apoptosis induction. Dephosphorylation of Bcl2-associated agonist of cell death protein was increased after methoxyphenyl chalcone treatment that led to activation of caspases. Interestingly, this drug also worked as a G2/M checkpoint modulator with alternative ways of DNA damage signal–evoking potential that might work to increase response after cisplatin treatment. In addition, methoxyphenyl chalcone was able to suppress autophagic flux and stemness regulator in ovarian spheroids that decreased their survival. Therefore, combination of methoxyphenyl chalcone and cisplatin showed synergistic effects. Taken together, we believe that our novel compound is a promising novel therapeutic agent for effective clinical treatment of ovarian cancer.
Keywords
Introduction
Ovarian cancer is usually diagnosed in late stage and is one of the most significant cause of death from gynecologic malignancy in the world. From the latest global cancer statistics, it is estimated that 225,000 female were affected with 140,200 mortality rate annually. 1 The currently available chemotherapy following cytoreductive surgery gives valuable response in few months, but soon within the first year of completing the regimen of platinum/Taxol, about 75% of patients were relapse cases. 2 Therefore, development of a novel drug candidate for late stage ovarian cancer is demanding. In addition, a better treatment strategy to improve responsiveness against platinum-based therapy is also very important.
Mode of action of platinum-based therapy is mediated by interaction with DNA to form DNA adducts. Intrastrand DNA crosslink causes activation of several pathways that lead to DNA synthesis inhibition, cell cycle attenuation, and cellular death. In molecular basis, DNA adduct activates DNA damage signals, including activation of p53, p73, mitogen-activated protein kinase (MAPK), and ATR. 3 However, activation of those signaling can be attenuated by several events that cause cisplatin resistance. In late stage ovarian cancer, patients initially respond well to platinum-based therapy up to 70%; however, they soon gain resistance that causes 5-year survival rate below 20%. 2 Therefore, alternative ways to evoke DNA damage signal or any independent signals that promote cellular death are needed to improve responsiveness against cisplatin. From a genome-wide study, a number of genes were activated along with phosphorylation of Bcl2-associated agonist of cell death (BAD) protein that may correlate with acquired resistance of ovarian cancer against platinum. As a result, BAD phosphorylation prevents cisplatin-induced apoptosis. Patients with higher level of phosphorylated-BAD (p-BAD) had inferior overall survival rate than patients with lower p-BAD. 4 Thus, dephosphorylation or inhibition of BAD could resolve cisplatin resistance in ovarian cancer. 5 In addition, recent evidence supports the role of cancer stem cells (CSCs) as the tumor initiator and also the source of relapse after chemotherapy treatment in various tumors.6,7 CSCs have higher autophagic flux that maintains the cells for high metabolic demands and also serves as pro-survival mechanism. In bladder CSCs, inhibition of high autophagy flux through small interfering RNA (siRNA) and pharmacologic treatment successfully decreased the cell survival and improved the response to chemotherapeutic drugs. 8 In ovarian cancer, ovarian CSCs are enriched in ovarian spheroids that are commonly found in ascites of late stage patients. Ovarian spheroids play putative roles in progression, metastasis, and drug resistance. 6 Therefore, autophagy in ovarian CSCs may also be another promising target of therapy.
Phytochemical chalcone is a bioflavonoid that can be found in vegetables and fruits. Chalcone-based derivatives have been yearly investigated to treat several diseases, including inflammatory diseases and cancer. Cytotoxicity of chalcone derivative was reported against several cancer types, including leukemia, lung, breast, colon, and ovary. 9 Chalcone is able to upregulate tumor suppressors through inhibition of deubiquitination enzymes. 10 In addition, cell cycle checkpoint regulators are also downregulated by chalcone to inhibit cell cycle progression. 11 Here, we developed a novel chalcone-based derivative, (E)-1-4(4-aminophenyl)-3-(2,4-methoxyphenyl)prop-2-en-1-one or methoxyphenyl chalcone (MTC) as drug candidate in ovarian cancer. In this study, we explored the cytotoxic activity of the compound on ovarian cancer cells and try to overcome resistance in the cisplatin-resistant cells. We found that ovarian cancer cell lines that were originated from clear cell carcinoma and serous adenocarcinoma type were responsive to MTC in a concentration-dependent manner. MTC was able to suppress cell cycle progression in G2/M checkpoint and induce cellular death. The process might be mediated by dephosphorylation of BAD. More importantly, MTC was able to suppress autophagy-related protein 5 (Atg-5) that is important for autophagosome elongation. Inhibition of autophagy in ovarian spheroids successfully decreased their survival. Therefore, application of MTC had beneficial effects in combination with cisplatin. Taken together, we believe that our novel compound is a promising novel drug candidate to treat ovarian cancer, as well as to be an adjunct drug to improve treatment outcome in cisplatin treatment.
Materials and methods
Compound synthesis
The mixture of 4′-aminoacetophenon (6 mmol) and 2,4-dimethoxybenzaldehyde (6 mmol) was dissolved in 30 mL ethanol, and then 6 mL of NaOH 40% solution was added dropwise, while the temperature was kept under 10°C. The reaction mixture was stirred under this condition for 1 h. Stirring was continued at room temperature for 4 h. Thereafter, the reaction mixture was poured into ice-water. The precipitated solid was filtered off and recrystallized on aqueous ethanol. Purity of the compound was clarified with melting point analysis, and the molecular structure was confirmed with infrared (IR), nuclear magnetic resonance (NMR), and mass spectrometry (Bruker Daltonik GmbH, Germany).
Cell culture
Human ovarian cancer cell lines (ES-2 and Hey-A8) were received from American Type Culture Collection (ATCC, USA). Cells were cultured in McCoy’s 5A medium (Gibco, USA) supplemented with 1% antibiotic/antimycotic (#15140; Gibco) and 10% fetal bovine serum (FBS; Sigma-Aldrich, USA). Cells were maintained in a standard incubator (Shel Lab; Sheldon Manufacturing, USA) at 37°C and 5% CO2.
Spheroids formation
Ovarian spheroids were developed from ES-2 cells following the previously described protocol. 12 Parental cells, as much as 105, were seeded on non-attached flask with stem cell medium (Nutristem-XF; Biological Industries, Israel) that contains suitable growth factors for CSC enrichment at 37°C in 5% CO2 incubator for 2 days when the spheroids were clearly visible under microscope. Non–stem cells died due to starvation while CSCs kept alive and compacted to form spheroids. In order to get higher purity of CSCs, at least three passages were done prior to studies.
Cell viability assay
Cell viability for adherent cells against drugs was quantified with sulforhodamine B (SRB; Sigma-Aldrich). Cells were seeded on 96-well plates (at a density of 3500 cells/well) in 100 µL of McCoy’s 5A medium and left to attach for 24 h. Cells were treated with cisplatin (Abiplatin injection; Pharmachemie BV, The Netherlands) or MTC. Cells were washed with phosphate-buffered saline (PBS) and then fixed with 10% cold trichloroacetic acid for 1 h prior to incubation with SRB for 1 h at room temperature. Attached SRB dye on the viable cells was dissolved in weak base (20 mM Tris base) and read under an absorbance of 595 nm.
Western blot
Cells were washed with PBS twice and then lysed with ice-cold lysis buffer (radioimmunoprecipitation assay (RIPA) buffer; R0278; Sigma-Aldrich, USA). The protein lysate was collected after centrifugation to remove cellular debris on the pellet. The protein concentration then was quantified using bicinchoninic acid (BCA) assay kit (Pierce, Thermo Scientific, USA). The equal amount of protein lysate from each sample was run on 10% sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE), followed by protein transfer to polyvinylidene difluoride (PVDF) membrane. Primary antibodies originated from mouse or rabbit were used to probe the protein level from each well (the list of antibodies was presented in Supplemental Table 1). After overnight probing, the interaction of protein–primary antibody was identified using horseradish peroxidase (HRP)-linked secondary antibody. The signal was detected using UVP imaging system (UVP, USA).
Immunofluorescence staining
For adherent cells, cells were seeded on 24-well plates with a coverslip on the bottom of each well. After treatment, cells were washed twice with PBS, fixed with 4% formaldehyde, and probed with primary antibody (cleaved-poly(ADP-ribose) polymerase (PARP), p21/Clip1, or c-Myc) at 4°C for overnight. Fluorophore-conjugated antibody (Alexa Fluor; Life Technologies, USA) was used to track the in situ interaction of protein–primary antibody. Double-stranded DNA staining (4′,6-diamidino-2-phenylindole (DAPI); Invitrogen, USA) was used as nuclear staining. The fluorescence signal was captured under confocal microscopy (Nikon, Japan).
Apoptosis analysis
Annexin V–propidium iodide (PI) double staining was performed with flow cytometry to quantify the percentage of apoptotic cells in each sample. Cells (106 cells for each sample) were treated with Annexin V and PI at room temperature for 15 min in the dark. The samples were immediately analyzed using flow cytometry (LSRFortessa; BD Biosciences, USA).
GFP-LC3 transfection
GFP-LC3 was transfected to ES-2 cells to study the autophagosome formation and function in ovarian cancer, especially in response to MTC treatment. Two millions of ES-2 cells were mixed with 10 µM GFP-LC3, and transfection was performed with the electroporation technique following the manufacturer’s protocol (Invitrogen).
Cell cycle analysis
Cell cycle was analyzed with the properties of PI that is able to analyze cellular DNA content. By this method, three major cellular cycle phases can be detected, including G1, S, and G2/M. Samples (105 cells per sample) were pretreated with PI for 30 min prior to analysis with flow cytometry (LSRFortessa; BD Biosciences).
Trans-well migration assay
ES-2 cells were resuspended in serum-free McCoy’s 5A medium and placed in trans-well chamber with or without MTC administration. After 18 h, migrated cells were fixed and then stained with Giemsa.
Combination analysis
Combination of cisplatin and MTC was analyzed using Chou-Talalay algorithm of multi-drug combination. CompuSyn software (ComboSyn Inc., USA) was used following the guideline of two drug combination analysis. Combination index (CI) was used to determine whether both drugs have synergistic, additive, or antagonistic effects.
Statistical analysis
Statistical analysis was performed using GraphPad Prism statistical tools (GraphPad Software, Inc., USA). In the descriptive analysis, measures of central tendency (mean and median) and dispersion (standard deviation) were determined. Level of confidence was set at 95%.
Results
MTC has cytotoxic effects on ovarian cancer cells
As a result of the synthesis process, (E)-1-4(4-aminophenyl)-3-(2,4-methoxyphenyl)prop-2-en-1-one or MTC that is a chalcone-based derivative was generated. MTC has calculated molecular weight of 284.1287 following our confirmatory findings by mass spectrometry (Figure 1(a)). Molecular structure of MTC was represented in Figure 1(b).
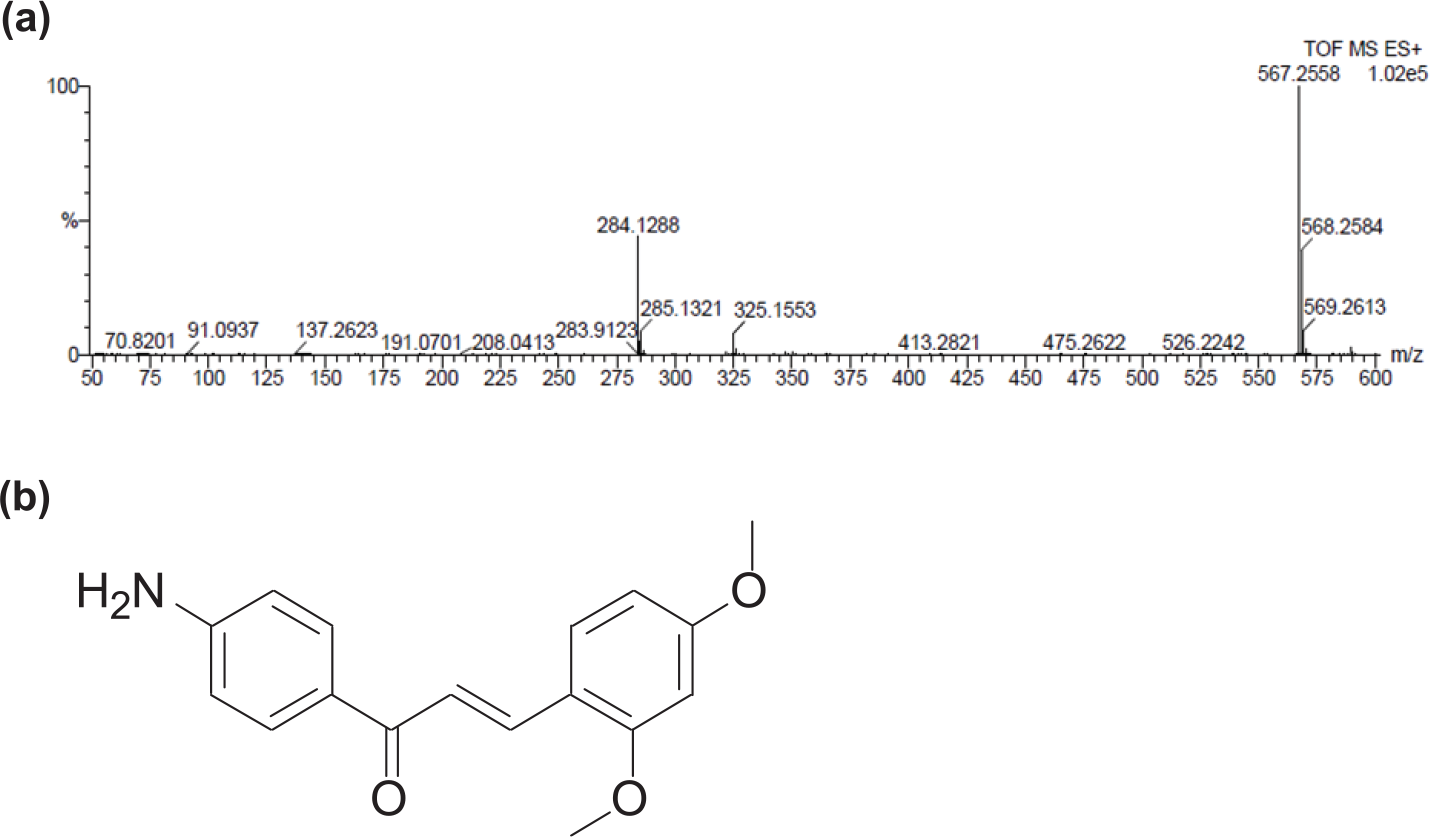
Chemical structure and molecular weight of chalcone derivative MTC. (a) The molecular mass and formula of chalcone derivative was assessed with mass spectrometry. Calculated molecular mass was 284.1287 with formula of C17H18NO3. (b) Molecular structure of (E)-1-4(4-aminophenyl)-3-(2,4-methoxyphenyl)prop-2-en-1-one (MTC) was presented.
For the detection of cytotoxic activity in ovarian cancer cells, SRB assay was used to detect the viable cells. Ovarian cancer cells were treated with MTC in various concentrations for 24 and 48 h. Viable cells were then quantified with SRB assay. Figure 2 clearly show the concentration-dependent effects of MTC on ovarian cancer cells. We found that MTC has potential therapeutic effects to eliminate ovarian cancer cells. Two ovarian cancer cell lines from different subtypes were used in this study. ES-2 was originated from high-grade clear cell carcinoma. This subtype was mentioned as one of the poor prognosis predictors because the patients have poorer outcome and high resistance to established platinum-based chemotherapy. 13 In this study, we also used the most prevalent subtype, Hey-A8, that originated from late stage serous cystadenocarcinoma. Serous type adenocarcinoma is the most common type of ovarian cancer 14 ; thus, cytotoxic activity against this cell type gives opportunity to be translated into a majority of patients.

MTC has cytotoxic effects on various subtypes of ovarian cancer. Cytotoxicity of MTC was assessed in two ovarian cancer cell lines: ES-2 and Hey-A8. Cells were treated with MTC with concentrations ranging from 6.25 to 100 µM for 24 or 48 h. Viability of survived cells after treatment was quantified with sulforhodamine B assay.
Apoptosis induction by MTC is mediated by activation of DNA damage signals through p-BAD dephosphorylation
Photomicrographs were taken after 48 h of treatment with MTC (Figure 3(a)). Then, cells were fixed with 4% formaldehyde and probed with antibody against cleaved-PARP for immunofluorescent staining. Since PARP is the major substrate of caspase, cleaved-PARP accumulation could indicate higher activity of caspase. In order to quantify the proportion of apoptotic cells in each sample, Annexin V–PI double staining was performed with flow cytometry. Anticoagulant protein Annexin V could bind exposed phosphatidylserine that is exposed on the surface of cell membrane due to asymmetry in the early phase of apoptosis. Thereby, fluorescein isothiocyanate (FITC)-conjugated Annexin V is able to detect the early phase of apoptosis. Only cells with co-staining with PI indicate chromatin condensation, the late stage process of apoptosis. The percentage of cells with Annexin V and PI staining was compared with control with flow cytometry. We found that application of MTC could promote cellular death via apoptosis mechanism. The proportion of apoptotic cells increased along with higher concentration given (Figure 3(b)). Several proteins that are important in apoptosis regulation were clarified with western blotting (Figure 3(c)). From the western blotting, the apoptosis pathway was activated after MTC treatment. We checked protein level of p-BAD, cleaved caspase-9, and cleaved caspase-7. We found that caspase was activated to promote apoptosis. This caspase activation was consistent with BAD dephosphorylation.

MTC induces apoptosis in ovarian cancer cells. (a) Photomicrographs of ES-2 cells treated with MTC for 24 h showed the concentration-dependent effect on cytotoxicity. Immunofluorescent staining to identify cleaved-PARP was performed to represent caspase activity. Cleaved-PARP as the main substrate of caspase-3 was found abundant after MTC treatment, indicating ongoing apoptosis. (b) After treatment with MTC for 24 h, quantitative proportions of early and late apoptotic phases of ES-2 cells were clarified with Annexin V/PI double staining. (c) Western blotting was performed to study the regulation of apoptosis after MTC treatment for 24 h. Status of BAD phosphorylation and activation of caspases were clarified. Apoptosis-inducing effect of MTC was found consistent with dephosphorylation of p-Bad. As a result, caspase activation was present to allow apoptosis.
MTC inhibits cell cycle progression in G2/M phase
Cellular status in cell cycle was also studied following MTC treatment. After 48 h of treatment, ES-2 cells were analyzed for their cell cycle status with flow cytometry and their cell lysates were assessed for cyclin expression with western blotting. Immunofluorescent staining to identify p21WAFI/CIP1 and c-Myc was also performed. From western blotting, cyclin B1 was suppressed by MTC treatment (Figure 4(a)). We found that compared with controls, cells that were treated with MTC exhibited decreased cell cycle progression. Proportion of cells in G2/M phase was increased significantly (G2/M arrest) after treatment (Figure 4(b)). G2/M arrest is also known as DNA damage checkpoint that delays or arrests cell cycle progression in response to DNA damage. The promotion of G2/M arrest was caused by inhibition of cyclin B1 activity after MTC treatment. The mechanism of cyclin B1 inhibition may be initiated by the cyclin-dependent kinase (CDK) inhibitor p21 (also known as p21WAFI/CIP1) upregulation. p21WAFI/CIP1 is a notable CDK inhibitor that acts as both sensor and effector of certain anti-proliferative stimulus, including DNA damage signals. 15 p21WAFI/CIP1 could inhibit cyclin B1/cdc2 activity that acts as key regulator in G2/M phase checkpoint. Therefore, we also observed the upregulation of p21WAFI/CIP1 after MTC treatment (Figure 4(c)). In contrast with the fluorescent in situ imaging, we also found that expression of c-Myc was downregulated.
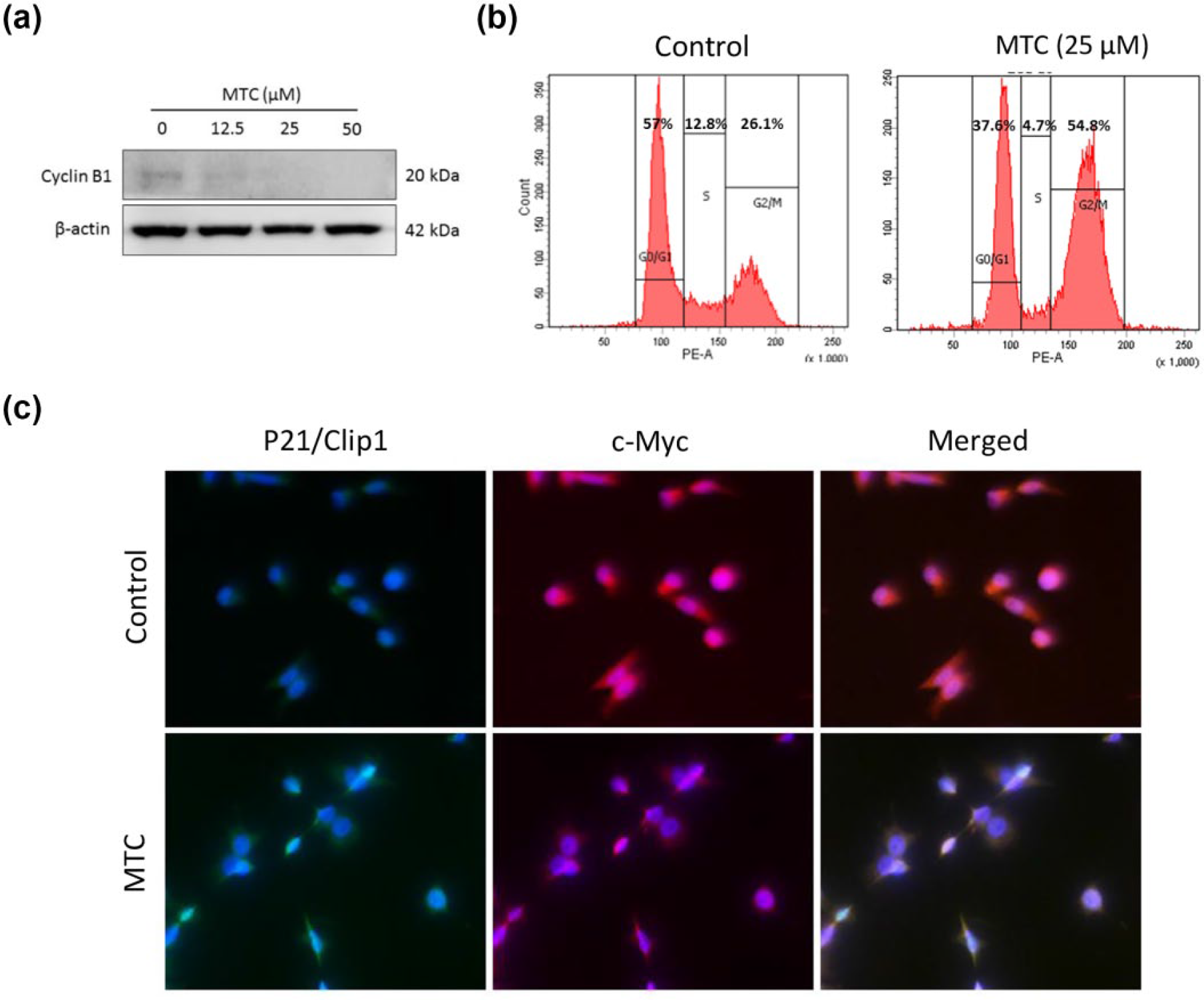
MTC inhibits cell cycle progression. (a) After treatment with MTC at different concentrations for 24 h, expression level of cyclin B1 in ES-2 cells was analyzed with western blotting. (b) Flow cytometry was performed to study the status of ES-2 cells in cell cycle. Cells that were treated with 25 µM of MTC for 24 h were compared with controls. (c) From immunochemistry staining, p21 as tumor suppressor was activated in ES-2 cells treated with MTC (25 µM) for 24 h. In contrast, c-Myc was found suppressed after MTC treatment.
Autophagy suppression by MTC decreased survival of ovarian spheroids through Sox-2 downregulation
Autophagy becomes important for cancer cells to meet the high metabolic demand associated with rapid proliferation or as a survival mechanism to protect their life under stress. Autophagy-related stress tolerance was found to promote cell survival by maintaining energy production that can lead to tumor growth and chemoresistance. 16 We initially transfected GFP-LC3 into ES-2 cells to study the autophagosome formation during the autophagy process. Then, autophagy-related proteins were clarified with western blotting following MTC treatment. Suppression of autophagosome formation by MTC was seen from photomicrographs with fluorescent microscopy. Autophagy is highly modulated by autophagy-related genes, and Atg-5, in particular, plays a major role in autophagy regulation through promoting autophagosome elongation. 17 We found that after MTC treatment, Atg-5 was downregulated that caused inhibition of mature autophagosome, as we can see from inhibition of lipidated LC3. As a result, caspase-7 was activated (Figure 5(a)). Therefore, because of Atg-5 suppression, autophagy inhibition was seen under a fluorescent microscope (Figure 5(b)). We then developed ovarian spheroids from ES-2/GFP-LC3 cells following the previously described protocol. 12 Spheroid formation ability is one of the CSC characters via self-renewal property, so it can also be used to enrich CSCs. 18 After 24 h of treatment with 25 and 50 µM MTC, ovarian spheroids were disrupted and the viability was significantly decreased (Figure 5(c) and (d)). From the three-dimensional immunofluorescence of ovarian spheroids, inhibition of LC3 was consistent with Sox-2 suppression (Figure 5(e)). Sox-2 is one of the key regulators for ovarian CSCs regulation. 6 In addition to MTC inhibitory effect on Sox-2, in a confirmatory assay, using western blot, the ovarian spheroids when treated with different concentrations of MTC exhibited a dose-dependent significant downregulation of the pluripotent transcription factors, Oct4, KLF4, and c-Myc (Figure 5(f)). Therefore, elimination of ovarian spheroids by MTC treatment might be mediated by autophagy inhibition.

MTC has cytotoxic effects on ovarian spheroids through autophagy inhibition and suppression of Sox-2. (a) Expression levels of Atg-5, LC3, and cleaved caspase-7 in ES-2 cells treated with MTC (25 µM) for 24 h were assessed by western blotting. β-actin was used as internal control in control and treated samples. (b) After 24 h of treatment, ES-2 cells that have been transfected with GFP-LC3 were fixed and assessed under a fluorescent microscope. Nuclear staining was done with administration of DAPI. Red arrow indicates mature autophagosome, while white arrow represents autophagosome puncta. (c) Ovarian spheroid was developed from ES-2/GFP-LC3 with stem cell medium. Then, cytotoxic effect of MTC to ovarian spheroids was seen under a light microscope. (d) Quantification of viable cells from spheroids was analyzed with Alamar blue assay (mean ± SD). (e) Three-dimensional immunofluorescent staining was performed to see the in situ expression of Sox-2, in colocalization with LC3. DAPI was used to stain nucleus. **p < 0.05. (f) Ovarian spheroids when treated with different concentrations of MTC exhibited a dose-dependent significant downregulation of the pluripotent transcription factors, Oct4, KLF4, and c-Myc.
MTC suppresses the migration ability of ovarian cancer cells in vitro
There is a significant positive correlation between metastatic status and prognosis among ovarian cancer patients, as the formation of metastases in ascites is a typical features of late stage ovarian cancer. Therefore, inhibition of metastases is very important to improve clinical outcome. N-cadherin and c-Met are essential for metastasis and progression in various tumors.19,20 We sought to study the suppression of N-cadherin and c-Met in correlation with the migration ability of ovarian cancer cells after MTC treatment. N-cadherin and c-Met expression profiles were evaluated with western blotting, while the migration ability of ES-2 cells was analyzed with trans-well migration assay. We found that N-cadherin and c-Met were suppressed by MTC administration (Figure 6(a)). MTC treatment could successfully suppress migration ability in vitro (Figure 6(b)).
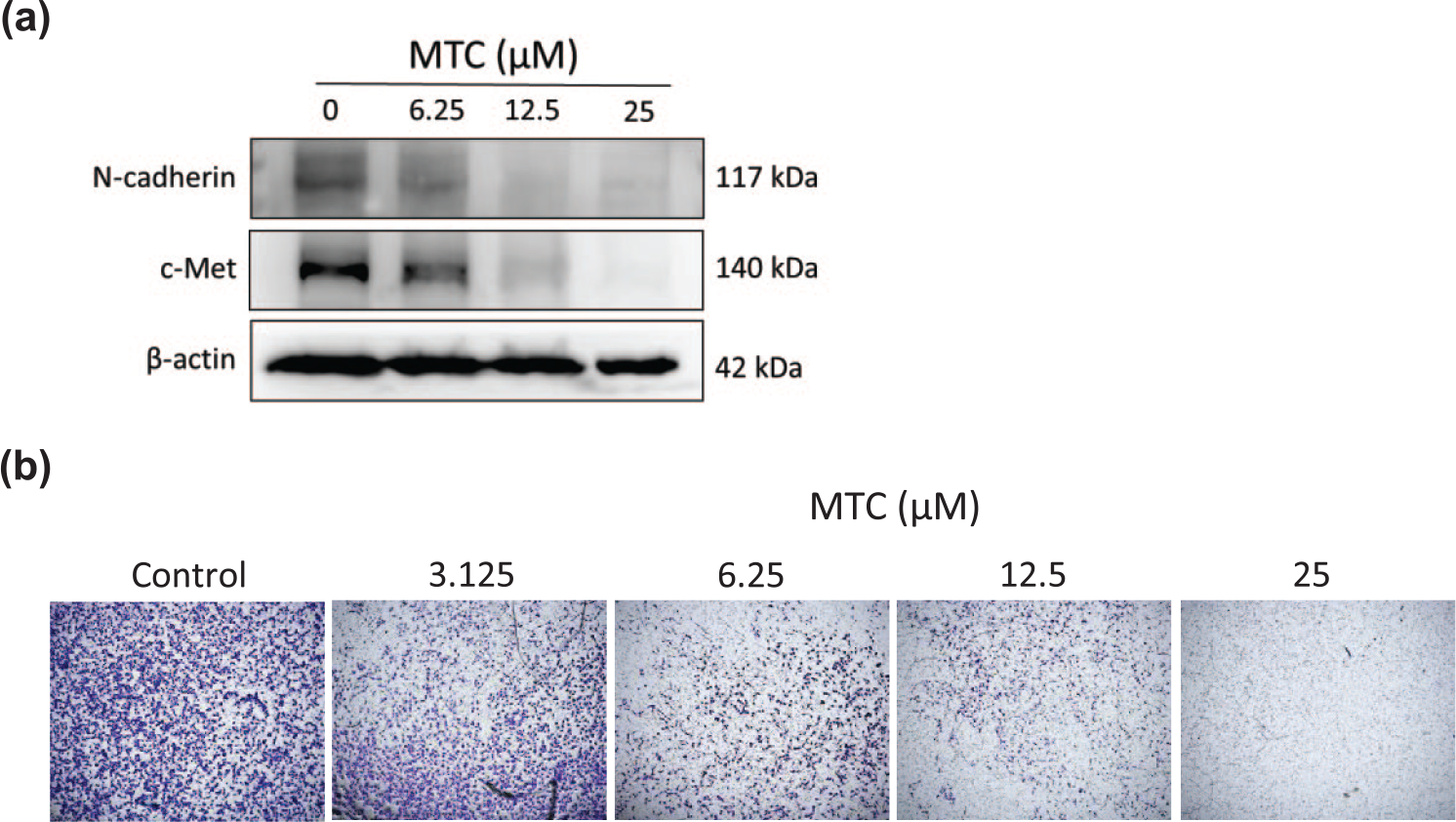
MTC inhibits ovarian cancer cell migration ability through N-cadherin and c-Met suppression. (a) In order to study suppression of key regulators for invasiveness and metastases by MTC treatment, western blotting was performed with antibody to probe N-cadherin and c-Met. (b) With lower concentration, starting from 6.25 µM for 8 h, ES-2 cells migration was assessed in trans-well chambers. Images were captured after cellular staining with Giemsa.
MTC enhances the anti-cancer effect of cisplatin
We co-administered MTC with cisplatin using various concentration regimens (Figure 7(a)). After 24 h of treatment, we found that combination of MTC and cisplatin had synergistic cytotoxicity on ES-2 cells (Figure 7(b)). This observed synergistic effect was corroborated by data obtained from the Chou-Talalay drug combination algorithm using the CompuSyn software. Synergism is defined with combination index (CI) < 1, additive effect is achieved when CI = 1, and antagonism is inferred when CI > 1. Synergism was seen as dots inside the triangle. This demonstrated synergism is indicative of the potential role of MTC as an adjunct to cisplatin.
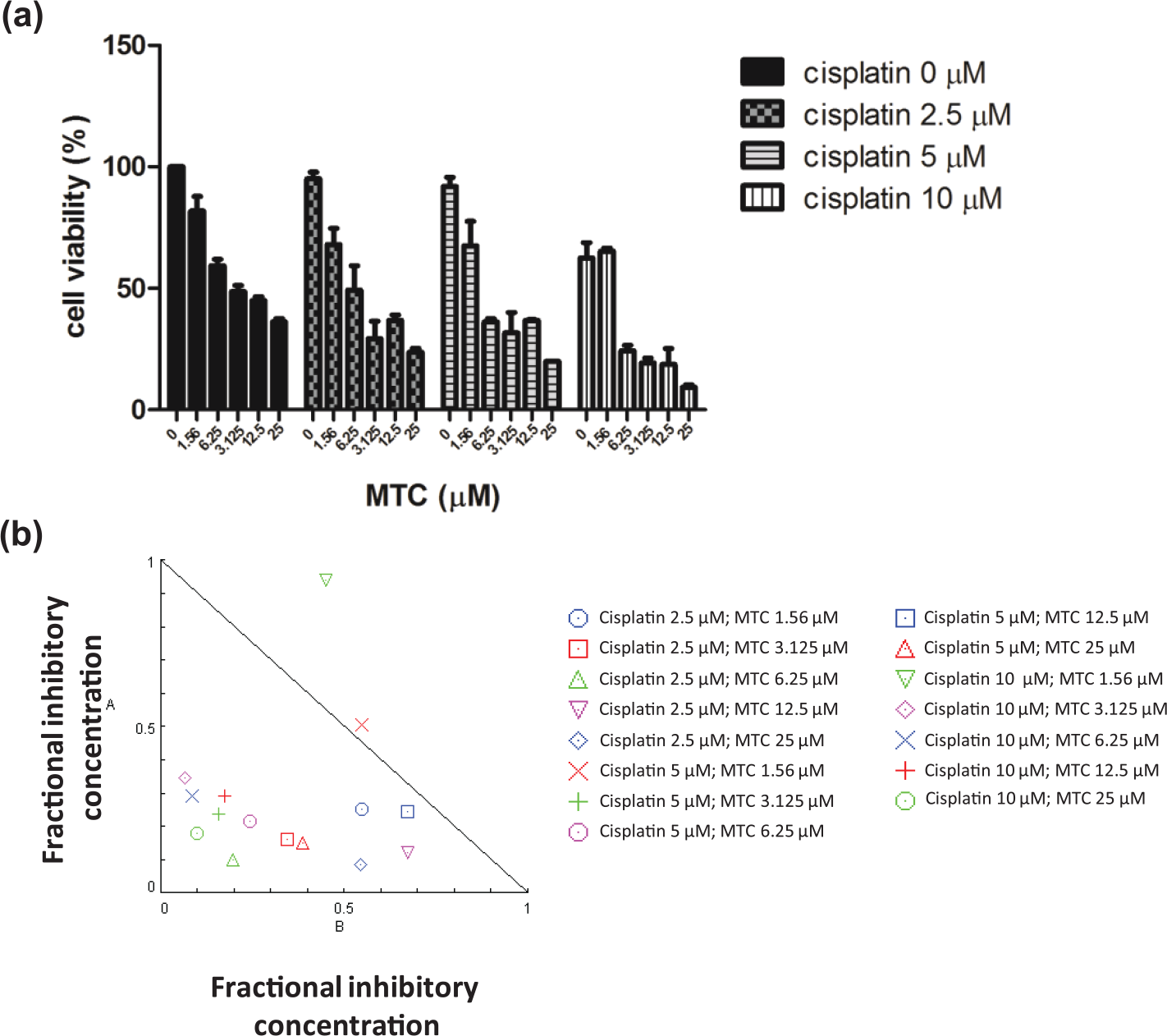
Combination analysis of MTC and cisplatin shows synergistic effect. (a) ES-2 cells were treated with monotherapy of cisplatin or combination of MTC and cisplatin. Cell viability was then assessed with SRB assay. (b) Combinational effects of cisplatin and MTC were analyzed with CompuSyn software. Combination index (CI) was presented in a triangle diagram, where synergistic effects were represented as dots inside the triangle. We found that combination of MTC and cisplatin had synergistic effect with CI < 1.
Discussion
Ovarian cancer has a high mortality rate with considerable metastasis ability and resistance to established chemotherapy. Clear cell carcinoma subtype only represents 3.7%–12.1% of all epithelial ovarian cancer cases. 21 However, clear cell carcinoma patients in advanced stage have relatively poor prognosis with lower 3-year and 5-year survival rates compared with the non–clear cell carcinoma subtype.13,22 Late stage clear cell carcinoma patients also have higher tendency of platinum insensitivity. 23 In this study, we used ES-2 cell line which originated from late stage clear cell carcinoma patient and found that the cell line was responsive to MTC (Figure 2). This finding is suggestive of a potential curative role of MTC in the currently therapy-elusive late stage clear cell ovarian cancer patients. In addition, another cell line, Hey-A8, from late stage serous ovarian carcinoma. Serous ovarian adenocarcinoma is the most common subtype of ovarian cancer. Thus, the potency of MTC is not only limited to clear cell carcinoma but may also be beneficial for a majority of ovarian cancer patients.
While the late stage clear cell carcinoma is associated with poor prognosis due to enhanced resistance to platinum-based therapy, interestingly, early stage clear cell ovarian carcinoma is highly responsive to platinum-containing chemotherapy. 23 Therefore, ES-2 is an ideal model to explore and develop a therapeutic strategy against acquired resistance in ovarian cancer. Several mechanisms involved in platinum resistance in clear cell carcinoma have been proposed, including drug efflux mechanism, drug detoxification, and DNA repair mechanism. In addition, several mechanisms of cell survival have been reported to correlate with platinum resistance in clear cell carcinoma, such as phosphorylation of BAD, 4 and HER2/neu activity and/or expression. 24 We demonstrated that clear cell carcinoma cells were apoptotic following MTC treatment, and that the observed induction of apoptosis might be mediated by dephosphorylation of BAD protein. The inhibition of BAD phosphorylation was related to caspases activation that initiates apoptosis pathway (Figure 3). BAD is a proapoptotic protein that can inhibit the Bcl-2 anti-apoptotic protein, facilitate the activity of other proapoptotic proteins and activate cytochrome-c release to induce apoptosis. This proapoptotic property of BAD is regulated by its phosphorylation. Only unphosphorylated BAD could interact with anti-apoptotic Bcl-2 family members, while p-BAD is sequestered in the cytosol and remains inactive by binding to 14-3-3 proteins. 25 In ovarian cancer patients, p-BAD has been reported to be positively correlated with a lower overall survival rate cisplatin resistance. 4 By contrast, inhibition of BAD phosphorylation reverses the cisplatin resistance to improve treatment outcome. 5 In our present study, we found that BAD dephosphorylation level was consistent with caspase-9 and caspase-7 activation to modulate cellular death. PARP-1 plays a critical role in double-strand break (DSB) repair, including the DSB repair process after cisplatin treatment that leads to chemoresistance. Inhibition of PARP-1 was found to be essential to sensitize cancer cells to cisplatin. 26 However, PARP-1 is one of the essential proteins that are digested fast and early by caspases -7 and -9. We found that activation of caspases might cause PARP-1 cleavage as evidenced by increased cleaved-PARP expression after treatment with MTC (Figure 3(a)). This observed cleavage of PARP-1 by our novel drug, MTC, is indicative of its putative role as a potentiator of cisplatin.
Apoptosis induction by MTC seems to be mediated by DNA damage signals as cells were arrested in G2/M phase (Figure 4(b)). Single agent therapy with MTC enhanced the activity of CDK inhibitor p21WAFI/CIP1. Inhibition of cell cycle progression limits proliferation of cancer cells, however, the cell cycle arrest at G2/M phase also allows cells to repair the damage. PARP-1 is the main player in DNA repair machinery. In our study, despite the G2/M phase arrest-related DNA repair, we observed that PARP-1 is cleaved following MTC treatment. Thus, indicating a dysfunctional DNA damage repair mechanism upon exposure to MTC. In addition, we observed the upregulation of p21WAFI/CIP1 with associated c-Myc suppression after MTC treatment (Figure 4(c)). c-Myc is pivotal in cancer survival and progression. In breast and ovarian cancers, c-Myc overexpression is associated with poor prognosis through its induction of multi-drug resistance (MDR) genes. 27 In breast cancer, c-Myc signaling can be deregulated by endogenous p21WAFI/CIP1 upregulation which contributes to tumor suppression. 28 In addition, inhibition of c-Myc may also inhibit cell cycle progression 29 and decrease epithelial–mesenchymal transition (EMT) features and properties of CSCs in breast cancer. 28 In order to study the metastasis inhibition effect of MTC treatment, we treated ES-2 cells with a lower concentration of MTC and found that lower concentration of MTC treatment could limit metastases through inhibition of N-cadherin and c-Met pathway (Figure 6(a)).
Impaired regulation, activation, assembly, or function for autophagy has been increasingly studied, especially in correlation with chemotherapeutic response. Autophagy plays an important role in drug resistance of CSCs. A high autophagic flux is associated with CSCs and may protect them from stress and DNA damage. 8 Moreover, inhibition of autophagosome formation by gene silencing or co-treatment with autophagosome inhibitor successfully induced cellular death of pancreatic CSCs. 30 To study autophagy regulation by MTC in ovarian CSCs, we developed ovarian spheroids from a clear cell carcinoma cell line. Ovarian CSCs are commonly found in ascites fluid of late stage ovarian cancer patients. The spheroids are enriched with CSCs that are responsible for resistance to chemotherapeutic drugs, metastasis, and tumor relapse. 6 We showed that MTC treatment could decrease autophagosome elongation by Atg-5 suppression in ES-2/GFP-LC3 cells. MTC decreased autophagy in ovarian spheroids developed from ES-2/GFP-LC3. Suppression of autophagy was followed by Sox-2 suppression, altering CSC maintenance, as a result, ovarian spheroids viability was decreased after MTC treatment. Collectively, these data demonstrate that MTC inhibits cancer growth by inducing apoptosis through dephosphorylation of BAD, limiting cell cycle progression by upregulation of CDK inhibitor, blocking metastasis through inhibition of N-cadherin, and importantly decreasing ovarian spheroids viability through autophagy inhibition. Co-administration of cisplatin and MTC increased tumor cytotoxicity in a synergistic manner. A schematic summary of MTC mechanism of action is shown in Figure 8.
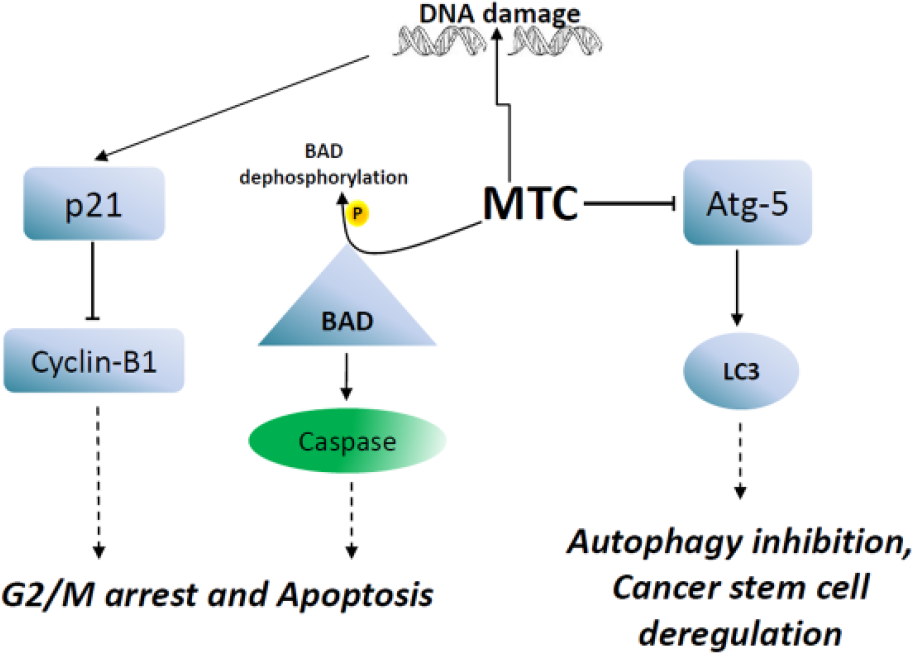
Methoxyphenyl chalcone (MTC) induces DNA damage and activates caspases through BAD dephosphorylation. Thus, MTC causes G2/M cell cycle arrest and apoptosis. In addition, MTC also suppresses Atg-5 that is important for autophagosome elongation and maturation. Autophagy inhibition may deregulate ovarian cancer stem cells.
Conclusion
We report a novel chalcone-based derivative, MTC, as a cytotoxic agent and a promising candidate for ovarian cancer therapeutics. We demonstrated that MTC exhibits tumor cytotoxic properties by apoptosis induction through p-BAD dephosphorylation. MTC also inhibit the autophagic activity of ovarian CSCs and thereby decreases their survival. This novel compound also suppress cell cycle progression and limit migration ability. More importantly, co-administration of MTC and cisplatin synergistically improve the cytotoxicity in ovarian cancer cells. This present study provides a basis for further exploration of the anti-CSC efficacy of MTC, singly or in combination with established chemotherapy, and is a first step towards establishing the clinical relevance of MTC in ovarian cancer patients.
Footnotes
Acknowledgements
This project was done under the collaboration of Taipei Medical University-Shuang Ho Hospital and Universitas Gadjah Mada. The authors thank Fang-Ting Kuo (Cell Sorter, Core Facility Center, Department of Medical Research and Education, Shuang Ho Hospital, Taipei Medical University) for her assistance with the flow cytometry, molecular, and cell-based assays. Y.K.S. and W.C.H. contributed equally to this work.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by National Science Council of Taiwan: W.C.H. (MOST-103-2113-M-324-001-MY2; MOST-103-2811-M-324-001) and W.H.L. (MOST-105-2320-B-038-054). This study was also supported by grants from Taipei Medical University (105TMU-SHH-15) to W.H.L. and grants from Taipei Medical University–National Taiwan University of Science and Technology Joint Research Program (TMU-NTUST-103-03) to C.T.Y.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.