
Abstract
Despite recent advances in diagnosis and therapy, prognosis of pancreatic cancer still remains very poor. Besides valid prognostic markers, novel therapeutic approaches are urgently needed. The family of cyclin-dependent kinases comprises 20 kinases which contribute to malignancy by promoting proliferation, migration, invasion, and apoptotic resistance of cancer cells. In this work, we investigated the role of CDK9 in pancreatic cancer. Immunohistochemical analysis of CDK9 expression in tumor and normal tissue of pancreatic cancer patients revealed an overexpression of CDK9 in pancreatic cancer tissue. In addition, high CDK9 expression in tumor tissue is associated with significantly shortened survival, especially in well-differentiated tumors. Moreover, the therapeutic potential of selective CDK9 inhibition on pancreatic cancer cells was evaluated by analysis of cell viability, long-term survival, and induction of apoptosis and characterized by western blotting and flow cytometry. Pharmacological CDK9 inhibition by SNS-032 drastically reduced cell viability in pancreatic cancer cells and potently suppressed long-term survival. Analyzing the mechanism of action revealed that CDK9 inhibition induced apoptosis and cell cycle arrest in a time-dependent manner by suppression of anti-apoptotic proteins. Furthermore, CDK9 inhibition potently enhances the therapeutic effect of chemotherapeutics in pancreatic cancer cells. In conclusion, we identified CDK9 as a negative prognostic marker in pancreatic cancer. Furthermore, pharmacological CDK9 inhibition is a novel and promising therapeutic approach for pancreatic cancer.
Keywords
Introduction
Today, pancreatic cancer is a major health problem evidenced by the fact that it is the fourth most common cause of cancer-related death worldwide counting 367,000 new cases diagnosed in 2015. Moreover, among all major cancers, pancreatic cancer has the highest mortality rate with its incidence almost equaling its mortality. 1 Despite recent progress in diagnosis and therapy within the last decades, prognosis of pancreatic cancer patients remains poor with a 5-year overall survival rate below 7%. 2 This can be attributed to the aggressive phenotype of pancreatic cancer cells with high invasive and metastatic potential. The only chance for cure is provided by a complete surgical resection. 3 However, due to vague and unspecific symptoms pancreatic cancer is in most cases only diagnosed at a locally advanced and/or metastatic tumor stage. For these patients, treatment remains palliative to alleviate symptoms and possibly limit cancer progression. 2 Due to their oncogenic background, pancre-atic cancer cells harbor resistance to conventional chemo- and radiotherapy, largely limiting its therapeutic efficiency. In addition, surgically treated patients with curative attempt often develop local tumor recurrence and/or metachronous metastases hampering the chance for cure and long-term survival. 4 Therefore, novel and reliable prognostic markers for early-stage pancreatic cancer are urgently needed to select patients who might take benefit from aggressive therapeutic approaches. Moreover, novel therapeutic approaches are warranted which exhibit anti-tumor potential in chemoresistant pancreatic cancers.
Members of the cyclin-dependent kinase (CDK) family play a crucial role in regulating the cell cycle. Besides, deregulation of CDKs and CDK-mediated pathways has been attributed to tumorigenesis and progression of different human cancer entities.5–8 Therefore, pharmacological inhibition of CDKs has been considered as a promising therapeutic approach for treating human malignancies. Consequently, over the past decade, research interests have focused on developing and evaluating CDK inhibitors to suppress exacerbated cell cycle progression of cancer cells.7,9 In addition to their cell cycle–regulating function, a subset of CDKs, most importantly CDK7 and CDK9, has recently been identified to be involved in regulating RNA polymerase (RNA Pol) II–mediated transcription.10,11 The large subunit of RNA Pol II is initially phosphorylated at the serine-5 (Ser5) residue of the C-terminal domain (CTD) by transcription factor II comprising CDK7 and Cyclin H, thereby facilitating transcription initiation. Next, the transcription elongation factor b (p-TEFb) consisting of CDK9 and Cyclin T phosphorylates RNA Pol II at serine-2 (Ser2) of its heptapeptide (YSPTSPS) repeats in the CTD to facilitate transcriptional elongation.10,12–14 Repression of RNA Pol II by CDK9 inhibition has been shown to block transcriptional elongation leading to suppression of short-living anti-apoptotic proteins such as Mcl-1, thereby promoting the induction of apoptosis. 10 Several CDK inhibitors with different inhibitory profiles for the family of CDKs have been identified, with a subset of them already undergoing clinical testing. Among them, SNS-032 appears to be the most selective CDK9 inhibitor currently under clinical investigation.15,16 In vivo activity of SNS-032 was confirmed in various animal models, and a phase-I trial demonstrated that patients with advanced solid tumors tolerated SNS-032 administration intravenously and orally.16,17 In this study we investigated the role of CDK9 in ductal pancreatic cancer as a prognostic marker and its potential as a novel therapeutic target for pancreatic cancer. Compared to normal pancreatic tissue we found that CDK9 expression is significantly increased in tumor tissue. Furthermore, elevated CDK9 expression was associated with significantly shortened survival, identifying CDK9 as a novel promising prognostic marker for pancreatic cancer. Moreover, CDK9 inhibition by SNS-032 resulted in markedly decreased pancreatic cancer cell viability and profoundly enhanced the effect of chemotherapeutic agent. Thus, we provide evidence for pharmacological CDK9 inhibition being a novel, highly active, and promising therapeutic strategy for pancreatic cancer.
Materials and methods
Patients and tissue samples
We collected tissue specimens of patients diagnosed with pancreatic ductal adenocarcinoma (PDAC) an treated in the Department of General and Visceral Surgery of the Ulm University Hospital between September 2000 and April 2012. Our study includes tumor and normal tissue from 54 PDAC patients. Samples were collected immediately during surgical resection in accordance with informed consent obtained from all patients. Histopathological and clinical data including patients’ overall survival, age, sex, tumor differentiation (grading), T classification, lymph node invasion, distant metastases, and tumor stage (according to the Union for International Cancer Control (UICC))18,19 were obtained (Table 1). The study was performed with the permission of the independent local ethics of the University of Ulm (approvals 112/2003, 268/2008, and 235/2015).
Patients’ characteristics.

UICC: Union for International Cancer Control.
Immunohistochemistry
Formalin-fixed paraffin-embedded sections of normal and tumor tissues were used for immunohistochemical studies. Hematoxylin/eosin–stained specimens were used for evaluating tumor burden and tissue quality. Sections of 2 µm thickness were deparaffinized and rehydrated in xylene followed by rehydration via transfer through graded alcohols. Citrate buffer pH 6.0 (BioGenex, San Ramon, CA, USA) was used as retrieval reagent while heating slices in the microwave using a pressure cooker. Microwave setup was 450 W for 15 min followed by 270 W for 10 min. Peroxidase solution was used as blocking reagent (Dako, Glostrup, Denmark). CDK9 antibody was diluted 1:150 (Cell Signaling Technology, Danvers, MA, USA). N-Histofine® Simple Stain MAX PO rabbit (Nichirei Corporation, Tokyo, Japan) was used to detect antigen. Staining was visualized using 3,3′-diaminobenzidine (DAB) + chromogen (DakoCytomation, Glostrup, Denmark). Mayer’s Hemalaun (Merck KGaA, Darmstadt, Germany) was used for counterstaining. Evaluation of immunohistochemical staining was performed by two independent individuals. To assess CDK9 expression a score was calculated. Therefore, staining intensity was defined (0 = no staining, 1 = low intensity, 2 = moderate intensity, and 3 = high intensity) and 500 carcinoma cells of each tumor-tissue slide were counted. The number of cells of each staining intensity was multiplied by intensity value. Determined score range was 0–1500 for tumor tissue. Due to limited access to normal pancreatic tissue and exclusion of stromal tissue, 100 epithelial cells of each normal-tissue slide were counted and scored according to above-mentioned scheme. When comparing normal to tumor tissue, the tumor tissue score was divided by 5 to adjust to the score of normal tissue.
Cell lines
The human pancreatic cancer cell lines BxPC3, 20 Colo 357, 21 and Panc89, 22 kindly provided by Prof. Anna Trauzold (University of Kiel), were cultured in RPMI 1640 Medium (Gibco®, Thermo Fisher Scientific, Schwerte, Germany) containing 10% fetal calf serum (Biochrom AG, Merck Millipore, Darmstadt, Germany), 1% sodium pyruvate, 1% L-glutamine, and 1% penicillin/streptomycin (Thermo Fisher Scientific). The human pancreatic duct epithelium (HPDE) cell line HPDE6-E6E7-c7, 23 kindly provided by Ming-Sound Tsao (Toronto), was cultured in a 1:1 mixture of Keratinocyte SFM medium and RPMI 1640 Medium (GlutaMAX™; Gibco, Thermo Fisher Scientific) containing 0.025% bovine pituitary extract, 2.5 µg/L epidermal growth factor (Gibco, Thermo Fisher Scientific), 10% fetal calf serum (Biochrom AG, Merck Millipore), and 1% penicillin/streptomycin (Thermo Fisher Scientific).
Western blot analysis
Cells were lysed in lysis buffer (30 mM Tris-HCl, pH 7.4; 150 mM NaCl; 2 mM EDTA; 2 mM KCl; 10% glycerol; 1% Triton X-100; Protease Inhibitor Tablet (Pierce™, Thermo Fisher Scientific), Phosphatase Inhibitor Cocktail 2 (Sigma-Aldrich, St. Louis, MO, USA)). Proteins were separated by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to a nitrocellulose membrane. Anti-CDK9, anti-Cyclin T1, anti-Bid, anti-Bax, anti-Bak, anti-Bcl-xL, anti-Mcl-1, anti-cIAP1, anti-Survivin, anti-XIAP, anti-PARP, anti-cleaved PARP (1:1000; Cell Signaling Technology), anti-actin (1:10,000; Sigma-Aldrich), anti-RNA polymerase II total (1:2000), anti-RNA polymerase II pSer2 (1:5000), and anti-RNA polymerase II pSer5 (1:1000; Covance, Princeton, NJ, USA) were used as primary antibodies. Immunocomplexes were detected using peroxidase-conjugated anti-mouse IgM (1:10,000; SouthernBiotech, Birmingham, AL, USA), anti-mouse IgG (1:10,000), and anti-rabbit IgG (1:5000) (both purchased from Cell Signaling Technology) followed by chemiluminescence detection (Pierce ECL Western Blotting Solution; Thermo Fisher Scientific).
Cell viability
Cell viability was determined by conventional 3-(4,5-dimethythiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) assay using a standard protocol as described previously. 24 Combination therapy treatment was performed by coincubating Panc89 cells with 300 nM SNS-032 (Selleck Chemicals, Houston, TX, USA) and 15 µM Irinotecan (Fresenius Kabi, Bad Homburg, Germany) or Gemcitabine (Hexal, Holzkirchen, Germany) for 24, 48, and 72 h. Color change was determined by photometer using a wavelength of 570 nm. Obtained results were normalized and GraphPad Prism 6 software (GraphPad Software, San Diego, CA, USA) was used to generate graphs for cell survival. To analyze long-term survival, cells were seeded in six-well plates and incubated with mock (dimethyl sulfoxide (DMSO)) or 300 nM SNS-032 for 24, 48, or 72 h. After incubation, cells were washed and cultured with complete medium for a total of 7 days. Cells were washed twice with ice cold phosphate-buffered saline (PBS), fixed in 10% formaldehyde for 30 min at room temperature, and stained with crystal violet (Sigma-Aldrich®; 1% crystal violet in 50% ethanol).
Cell cycle analysis
Cell cycle analysis was performed using the BD Cycletest™ Plus DNA Reagent Kit (BD Biosciences, San Jose, CA, USA) according to the manufacturer’s protocol. Cell cycle profiles were obtained using a FACScan flow cytometer (Becton–Dickinson, Franklin Lakes, NJ, USA) and CellQuest software (BD Biosciences).
Statistical analysis
Data were analyzed using IBM SPSS statistics 20 (SPSS Inc., Chicago, IL, USA). Statistical significance between groups was determined using log-rank test for Kaplan–Meier analysis and Wilcoxon signed-rank test for group comparisons demonstrated by boxplot. The p values <0.05 were considered statistically significant.
Results
Patients’ characteristics
Firstly, we determined the relevance of CDK9 expression in human pancreatic cancer by comparing normal and tumor tissue samples of pancreatic cancer patients. The patient characteristics of our cohort are summarized in Table 1. In this study, a total of 54 patients (31 males, 23 females) with a median age of 69.17 years (range = 34.98–82.99) were analyzed. Among them, 34 patients were diagnosed with well-differentiated tumors (grades 1 and 2; 63%) and 20 with poorly differentiated tumors (grades 3 and 4; 37%). Whereas no tumor was classified as T1, 3 tumors were classified as T2 (5.6%), 49 tumors as T3 (90.7%), and 2 tumors as T4 (3.7%). In 43 patients lymph node metastases were evident (79.6%). However, only in seven patients distant metastases were detected (13%). Recapitulatory, only one patient was assigned to stage I according to the UICC classification (1.8%), whereas 44 patients were diagnosed at stage II (81.5%). Two patients belonged to UICC stage III (3.7%), and seven patients were diagnosed at UICC stage IV (13%). The mean overall survival of our cohort was 17.11 months, ranging from 1.35 to 125.49 months. The 2-year survival was approximately 40%, whereas after five years, a mere 14% were still alive.
CDK9 is overexpressed in pancreatic tumor tissue
In order to investigate the CDK9 expression pattern in normal and carcinogenic pancreatic tissues, we performed immunohistochemical stainings using a monoclonal CDK9 antibody on paraffin-embedded pancreatic tissue samples. We randomly chose 18 PDAC patients of our cohort and compared CDK9 expression levels in normal and tumor tissues. Strong immunoreactivity could be observed in tumor tissue (mean score = 119.85), revealing a significant difference (p = 0.003) compared to healthy tissue (mean score = 68.70; Figure 1(a) and (b)). In line with this finding, western blot analysis revealed a higher expression of CDK9 in pancreatic cancer cell lines compared to the non-cancerous pancreatic duct cell line HPDE (Figure 1(c)). Thus, CDK9 is overexpressed in pancreatic cancer tissue.

CDK9 expression in healthy and cancerous pancreatic tissue and cell lines. (a) Representative pictures of immunohistochemically CDK9 and HE stained human normal and PDAC tissue. (b) Comparing CDK9 expression levels in normal and tumor tissues of 18 PDAC patients revealed a significantly increased immunoreactivity in tumor tissue compared to normal pancreatic tissue (p = 0.003). Data was analyzed using IBM SPSS statistics 20. Statistical significance between groups was determined using Wilcoxon signed-rank test for group comparison demonstrated by boxplot. The p values < 0.05 were considered statistically significant. (c) Markedly increased CDK9 expression was also shown in different pancreatic cancer cell lines compared to a human non-cancerous pancreatic duct epithelium cell line HPDE6-E6E7-c7. Cells were lysed and subjected to western blotting. One representative of two independent experiments is shown.
CDK9 is a negative prognostic marker in pancreatic cancer
Next, we investigated the potential role of CDK9 as a prognostic factor in pancreatic cancer. Therefore, we divided the CDK9 immunohistochemical staining scores of the tumor samples into two subgroups, resulting in a low-expression group with scores ⩽860 and a high-expression group revealing scores >860. We found that patients of the high-expression group had a mean overall survival of 10.92 months, whereas patients of the low-expression group had a mean overall survival of 20.59 months. In order to investigate a potential impact of CDK9 expression on overall survival of PDAC patients, Kaplan–Meier survival estimations were generated by correlating low- and high-expression group with patients’ clinicopathological parameters listed in Table 2. Intriguingly, our results revealed that patients suffering from tumors expressing high CDK9 levels have significantly worse prognosis compared to patients with low-level CDK9 expressing tumors (p = 0.009; Figure 2(a)). Representative examples of low- and high-level expressing tumors are shown in Figure 2(b). Interestingly, results become even clearer when subdividing our cohort by tumor differentiation (grading), revealing strong correlation between high CDK9 expression levels and decreased overall survival of PDAC patients with well-differentiated (grades 1 and 2) tumors (p < 0.001; Figure 2(c)). In contrast, patients with poorly differentiated tumors (grades 3 and 4) revealed no significant correlation of CDK9 expression and overall survival rates (p = 0.850; Figure not shown). Although no differences in the median CDK9 expression levels in lymph node negative and lymph node positive tumor tissue could be observed, patients with lymph node metastases revealed significantly increased survival rates when CDK9 expression levels were decreased (p < 0.001; Electronic Supplementary Material Figure S1). In summary, we identified CDK9 expression being a prognostic marker in pancreatic cancer for the first time.
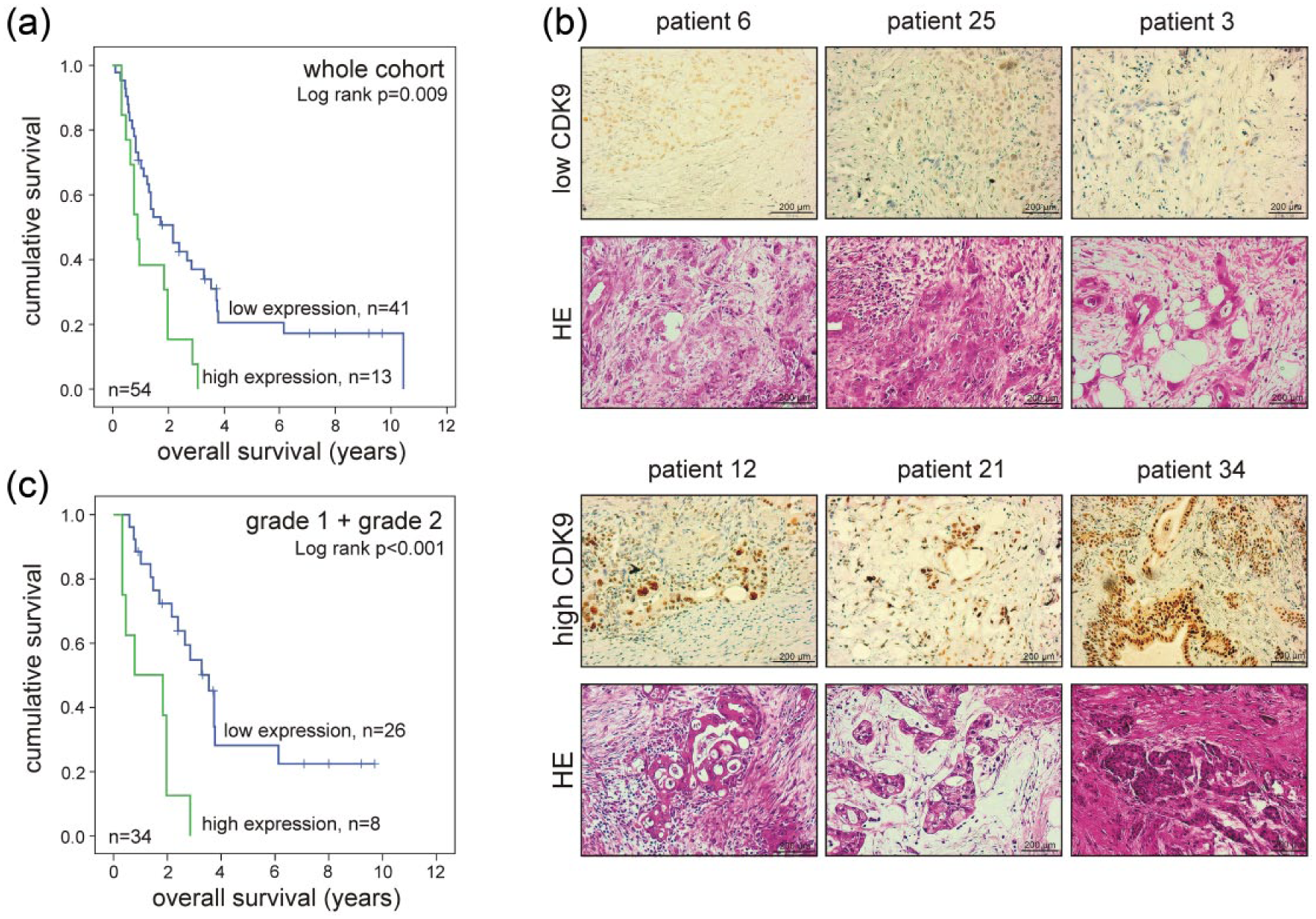
Impact of CDK9 expression on survival of PDAC patients. (a) Kaplan–Meier survival plot displaying the overall survival of the whole cohort according to CDK9 expression in pancreatic tumor tissue. Patients with tumors expressing low CDK9 levels show significantly (p = 0.009) increased survival rates compared to those with tumors expressing high CDK9 levels. (b) Representative pictures of immunohistochemically CDK9 and HE stained tumor sections of low CDK9 and high CDK9 expression group. (c) Kaplan–Meier survival plot displaying the overall survival of patients with well-differentiated cancers according to CDK9 expression. Patients with tumors expressing low CDK9 levels show significantly (p < 0.001) increased survival rates compared to those with tumors expressing high CDK9 levels. Data was analyzed using IBM SPSS statistics 20. Statistical significance between groups was determined using log-rank test. The p values < 0.05 were considered statistically significant.
Correlation of CDK9 expression with clinicopathologic indicators of PDAC.
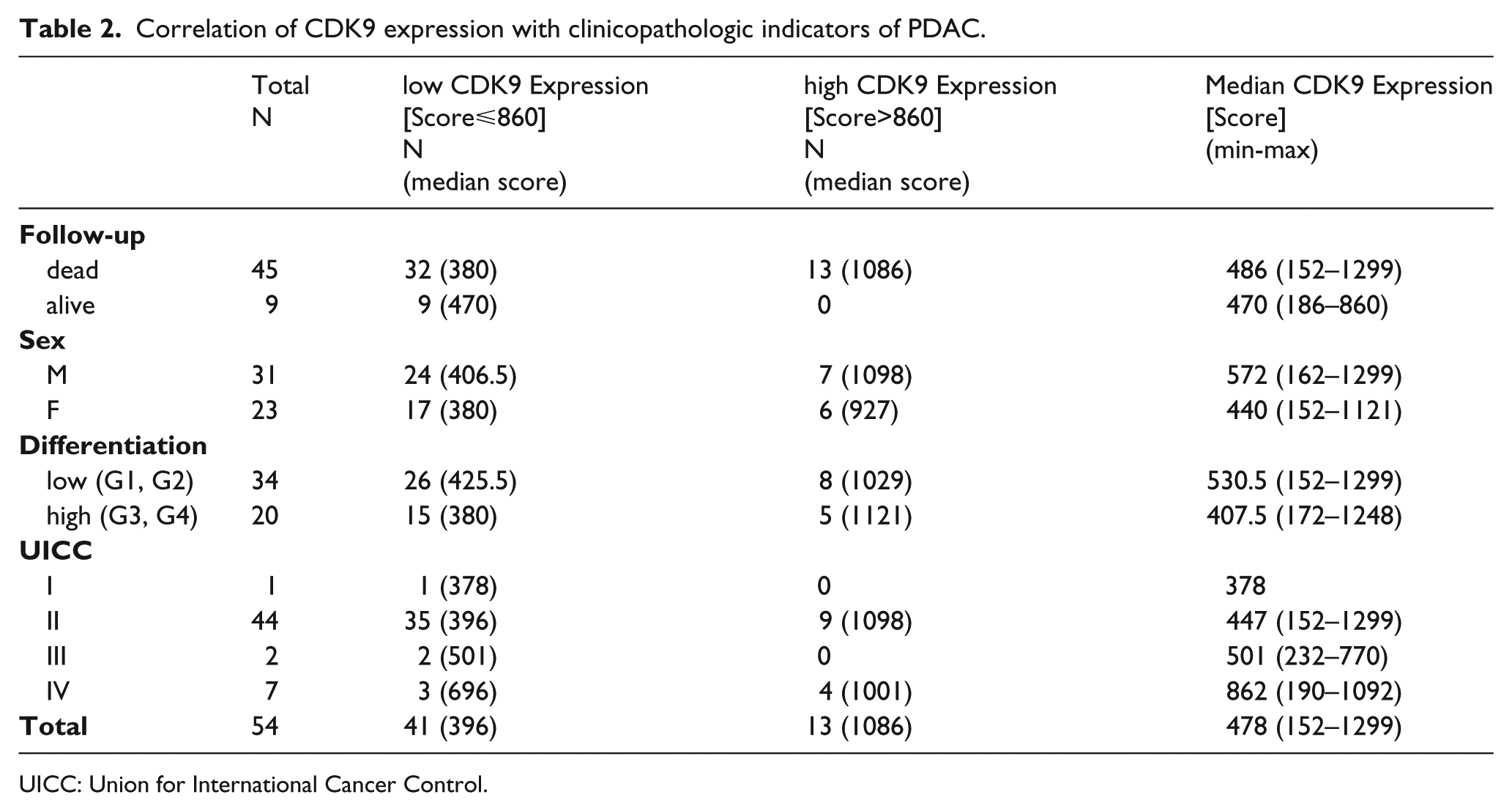
UICC: Union for International Cancer Control.
CDK9 inhibition by SNS-032 leads to decreased protein levels of CDK9 targets
So far, we have demonstrated that CDK9 is highly expressed in pancreatic cancer tissue and that high CDK9 expression significantly correlates with poor prognosis in pancreatic cancer patients. Based on these findings and given CDK9’s important role in controlling transcriptional elongation, we hypothesized that CDK9 inhibition harbors the potential as a novel therapeutic target in pancreatic cancer. First, CDK9 target expression including total RNA Pol II and RNA Pol II pSer2 in the pancreatic cancer cell lines BxPC3, Colo 357, and Panc89 was confirmed performing western blot analysis (Figure 3(a)). SNS-032 has been shown to inhibit CDK2, CDK7, and CDK9 selectively within the kinome. Beyond that, SNS-032 is currently the most selective inhibitor for CDK9 in clinical studies, since its inhibitory potential is at least 10 fold selective for CDK9 over CDK2 and CDK7. 15 Pharmacological characteristics and safety of SNS-032 upon systemic application have already been confirmed in first clinical trials. 16 Therefore, we selected SNS-032 to investigate the therapeutic potential of selective CDK9 inhibition in pancreatic cancer applying a clinically relevant therapeutic agent. Intriguingly, treatment of Panc89 cells with 300 nM SNS-032, a concentration which has been shown to be reached and sustained in plasma of patients, 16 induced rapid and drastic suppression of RNA Pol II pSer2 mediated by the potent inhibition of the CDK9/Cyclin T complex (Figure 3(b)). Moreover, suppression of RNA Pol II pSer5 was observed in a time-dependent manner. Interestingly, we detected a slight reduction in the total level of RNA Pol II upon treatment with SNS-032 for 72 h. This has been observed before by other authors and is most likely due to prolonged inhibition of transcriptional elongation.25,26 In summary, exacerbated CDK9 in pancreatic cancer can be successfully suppressed by the clinically available CDK inhibitor SNS-032.
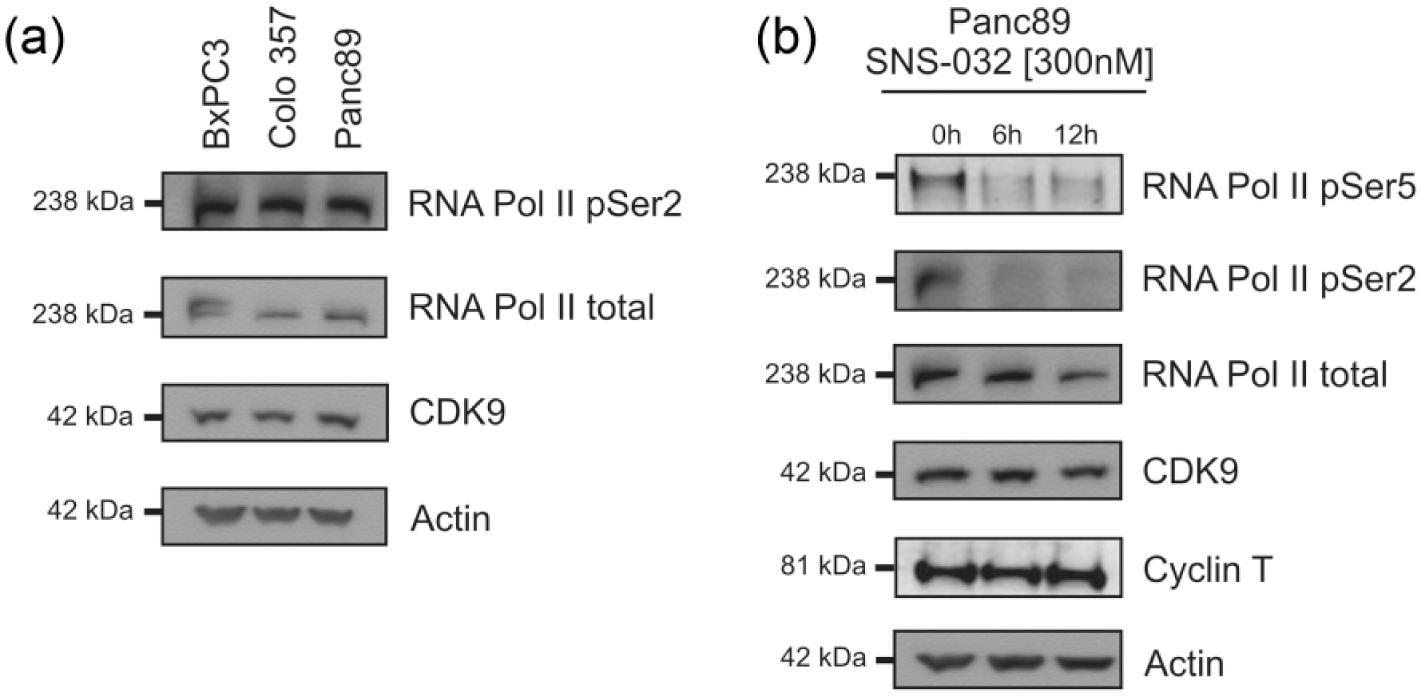
Expression of CDK9 target proteins and their reduction after SNS-032 treatment in Panc89 cells. (a) CDK9 target expression was confirmed in BxPC3, Colo 357, and Panc89 cells by subjecting cell lysates to western blotting. One representative of two independent experiments is shown. (b) Decreased CDK9 target expression after SNS-032 (300 nM) treatment was demonstrated in Panc89 cells for 6 and 12 h. Cell lysates were subjected to western blotting. One representative of two independent experiments is shown.
CDK9 inhibition by SNS-032 exhibits cytotoxic effects in pancreatic cancer cells by cell cycle arrest and apoptosis induction
Next, the cytotoxic effects of SNS-032 on BxPC3, Colo 357, and Panc89 cells were examined performing time course MTT assays. Treatment with 300 nM SNS-032 resulted in consistently and drastically reduced cell viability upon 24, 48, and 72 h (Figure 4(a)). While BxPc3 and Colo 357 cell growth is inhibited by approximately 50% and 40% after 72 h, respectively, Panc89 showed highest treatment response exhibiting a decrease in cell growth rate of 70%. Intriguingly, when examining long-term survival in Panc89 cells, we observed that SNS-032 treatment markedly reduced clonogenic survival upon 24 and 48 h and almost completely abrogated it after 72 h of treatment (Figure 4(b)). Cell cycle distribution analysis of Panc89 cells revealed a G2 cell cycle arrest upon 24 h treatment. Prolonged treatment with SNS-032 induced apoptotic cell death indicated by a drastic shift of cells to SubG1 phase after 48 and 72 h of SNS-032 treatment (Figure 5(a)). In summary, CDK9 inhibition by SNS-032 exhibits a potent anti-tumor effect in pancreatic cancer cells by a dual mechanism: inducing cell cycle arrest and apoptosis. To further elucidate the mechanism of apoptosis induction, we systematically analyzed the expression of important factors of the intrinsic apoptotic pathway (Figure 5(b)). Interestingly, we found the expression of pro-apoptotic proteins Bid and Bak remaining unchanged upon SNS-032 treatment, while loss of pro-apoptotic Bax could be observed. The latter has been observed before and can be explained by cellular mitochondrial translocation of Bax from a cytosolic to a membrane-bound form. 27 Importantly, a panel of anti-apoptotic proteins, including Mcl-1, Survivin, and XIAP, was suppressed upon SNS-032 treatment. The anti-apoptotic protein Bcl-2 could not be detected in Panc89 cells. In summary, apoptosis induction by CDK9 inhibition is mediated by a shift of the ratio of anti- and pro-apoptotic proteins facilitating Bax-/Bak-mediated apoptosis induction, confirmed by time-dependent increase of cleaved PARP. Taken together, these data illustrate dependence of Panc89 cells on CDK9 activity for survival by promoting the expression of anti-apoptotic proteins.

Impact of pharmacological CDK9 inhibition on pancreatic cancer cell viability. (a) Treatment with 300 nM SNS-032 resulted in consistently reduced cell viability upon 24, 48, and 72 h. Cell viability was determined by conventional MTT assay. Obtained results were normalized, and GraphPad Prism 6 software was used to generate graphs for cell survival. (b) SNS-032 treatment markedly reduced clonogenic survival of Panc89 cells when examining long-term survival. Cells were seeded in six-well plates, incubated with mock (DMSO) or 300 nM SNS-032 for 24, 48, or 72 h, and visualized after 7 days by crystal violet.

Changes in cell cycle distribution and induction of apoptosis in Panc89 cells under the influence of pharmacological CDK9 inhibition. (a) Cell cycle distribution analysis of Panc89 cells revealed an increased amount of G2 arrested cells after 24 h and a drastic increase of apoptotic cells after 48 and 72 h of SNS-032 treatment. Analysis of cell cycle distribution of Panc89 cells was performed by propidium iodide (PI) staining. Single cells were gated via PI width and area signals. Cell phases were calculated from a PI-area (FL2-A) histogram. Cell cycle profiles were obtained using a FACScan flow cytometer and CellQuest software. (b) Panc89 cells were treated with SNS-032 (300 nM) for the indicated times. Cells were lysed and subjected to western blotting in order to analyze various pro- and anti-apoptotic proteins. One representative of two independent experiments is shown.
Combination of CDK9 inhibition and chemotherapeutic agents synergize in killing pancreatic tumor cells
In order to investigate a potential additive effect of CDK9 inhibition in combination with a chemotherapeutic agent Panc89 cells were coincubated with 300 nM SNS-032 and 15 µM Gemcitabine or 300 nM SNS-032 and 15 µM Irinotecan. While treatment with SNS-032 in combination with Gemcitabine revealed only a moderate additive effect on Panc89 cell viability, Irinotecan potently synergizes with CDK9 inhibition. CDK9 inhibition by SNS-032 moderately synergizes with Gemcitabine in Panc89 cells leading to 15% reduced cell viability compared to single treatment (Figure 6(a)). Intriguingly, SNS-032 potently synergizes with Irinotecan decreasing cell viability by almost 40% compared to single treatment and almost completely abrogated cell survival of Panc89 cells (Figure 6(b)). In conclusion, CDK9 inhibitor–mediated anticancer activity can be substantially enhanced by the co-treatment with conventional chemotherapeutic substances.

Synergistic effect of SNS-032 treatment in combination with chemotherapeutic agents. (a) CDK9 inhibition by SNS-032 moderately synergizes with Gemcitabine in Panc89 cells. (b) SNS-032 potently synergizes with Irinotecan in Panc89 cells. Cell viability was determined after 24, 48, and 72 h by conventional MTT assay. Obtained results were normalized and GraphPad Prism 6 software was used to generate graphs for cell survival.
Discussion
Promising advances in diagnostics and therapy have been achieved for many cancer entities, paving the way for prolonged survival and cure for most cancer patients. In contrast, pancreatic cancer remains a mostly deadly disease and therefore a major challenge in clinical oncology due to its highly aggressive and invasive growth pattern and its tendency to early spread to distant sites. Conventional chemotherapy such as Gemcitabine remains the standard therapy for advanced pancreatic cancer. However, its therapeutic efficiency is largely limited by intrinsic or acquired chemoresistance of pancreatic cancer cells as well as therapy-associated morbidity, since conventional chemotherapy also targets healthy cells leading to therapy-associated toxicity. Consequently, the basis for successfully treating pancreatic cancer constitutes the identification of novel drug targets which are preferentially expressed in cancer cells and capable of translating cytotoxic effects in cancers while circumventing chemoresistance of these cells. In this study, we identified CDK9 being significantly overexpressed in pancreatic cancer tissue compared to normal tissue. Thus, CDK9 represents a potent drug target for selectively targeting cancer cells while sparing healthy, normal cells. Interestingly, some studies have suggested a role of deregulated CDK9-related pathways in tumorigenesis and tumor progression. A recent study has shown that CDK9 is highly expressed in head and neck cancer cell lines compared to non-transformed cells.28,29 However, for the first time, our study revealed CDK9 being overexpressed in malignant, primary tissue. These findings indicate that CDK9 has a central role in signaling pathways which determine malignant characteristics of cancer cells such as the ability to invade surrounding tissue, uncontrolled proliferation, and spread to other sites. 28 Indeed, it has been demonstrated that CDK9 mediates apoptotic resistance, a hallmark of cancer cells, by transcriptionally controlling the overexpression of anti-apoptotic proteins in cancer cells such as Mcl-1. 26 Furthermore, we demonstrate that CDK9 serves as a negative prognostic marker in pancreatic cancer. CDK9 is overexpressed in pancreatic cancer tissue of patients with a dismal prognosis. Based on these findings, it is tempting to speculate that CDK9 may contribute to the malignant behavior of a subset of even more aggressive cancers. In addition, these findings may indicate that CDK9 is highly expressed in a subset of patients in which conventional chemotherapy only achieved limited efficiency. Remarkably, our data revealed that the negative prognostic role of CDK9 is pronounced in well-differentiated pancreatic cancers. This may indicate that in de-differentiated cancers, an additional intracellular mechanism may control malignant characteristics which are controlled by CDK9 in well-differentiated cancers. Interestingly, in this context, a recent report has demonstrated that CDK9 expression is upregulated in the differentiation process of lymphomas. 30 Consequently, CDK9 represents a novel attractive drug target in pancreatic cancer and especially in patients with well-differentiated, chemoresistant pancreatic cancers, pharmacological CDK9 inhibition may be promising. This idea is supported by the finding that CDK9 inhibition is well tolerated in animal models and patients.16,17,31,32 However, so far, the comparison of CDK9 expression in healthy and malignant tissue was not provided. Initially, the role of CDKs was solely connected to controlling the cell cycle. Accordingly, inhibitors of different subsets of CDKs have been developed to counteract exacerbated cell cycle progression in cancer cells. 7 However, very recently, CDK9 was found to constitute the p-TEFb together with its partner Cyclin T to non-redundantly control transcriptional elongation.12,33 Intriguingly, the role of uncontrolled transcriptional elongation has long been known to be a key characteristic in cancer cells, proposing CDK9 as a promising therapeutic target. 34 In our study, we demonstrate that selective CDK9 inhibition is a powerful approach to induce cytotoxic activity in pancreatic cancer cells. We made use of SNS-032, the so far most selective CDK9 inhibitor currently in clinical study. Importantly, concentrations of the inhibitor which have been demonstrated to be reached and sustained in patients were sufficient to efficiently exhibit cytotoxic activity. 16 In this context, it is important to mention that the use of pharmacological inhibitors which are selective for CDK9 may be beneficial, since it is currently thought that the inhibition of the cell cycle–regulating CDK1 may lead to toxicity induced by unselective CDK inhibitors. 35 Mechanistically, we found that CDK9 inhibition caused apoptosis induction by the shift of ratio of anti-apoptotic and pro-apoptotic proteins in pancreatic cancer cells in line with previous reports that have shown that inhibition of transcriptional elongation may induce the suppression of short-living anti-apoptotic proteins. 10 Standard therapy for pancreatic cancer includes conventional chemotherapeutic approaches such as Gemcitabine and the FOLFIRINOX regime (comprising Irinotecan, 5-fluorouracil, folinic acid, and Oxaliplatin). Importantly, these therapeutic approaches revealed only limited efficiency in pancreatic cancer. This can be attributed to the oncogenic background of pancreatic cancer cells leading to intrinsic and/or acquired resistance. Thus, it will be decisive to identify novel strategies to overcome chemoresistance. Interestingly, our study demonstrates that the anticancer activity of standard therapeutic agents used in pancreatic cancer is substantially enhanced by CDK9 inhibition using SNS-032. Given its potency, the combination of SNS-032 and Irinotecan appears to be exceptionally promising, demanding further preclinical and possibly clinical evaluation. Thus, it will be exciting to determine the molecular mechanism of the mode of action of this novel combination. In this context, SNS-032 has already been reported to have radiosensitizing effects in lung cancer and increase the sensitivity of acute myeloid leukemia (AML) cells to Cytarabine.36,37 Furthermore, the CDK inhibitor dinaciclib has been described to synergize with cisplatin in preclinical models of ovarian cancer, 38 and dual inhibition of Cdc7 and CDK9 by PHA-767491 suppresses hepatocarcinoma synergistically with 5-fluorouracil. 39 These data support our finding that new treatment regimens combining CDK inhibitors with conventional treatment strategies are promising approaches in pancreatic cancer therapy.
Consequently, our study provides the basis for further preclinical and clinical evaluation of CDK9 inhibition in pancreatic cancer. Our data reveal that CDK9-induced activity is mediated by a dual mechanism, cell cycle arrest and apoptosis induction. Future studies will be required to further analyze the molecular basis of CDK9’s mode of action in pancreatic cancer cells. In conclusion, our results strongly demonstrates that CDK9 is a negative prognostic marker in well-differentiated, chemoresistant pancreatic cancer, and pharmacological CDK9 inhibition may present a novel approach in personalized pancreatic cancer therapy.
Footnotes
Electronic Supplementary Material
Acknowledgements
The authors thank Prof. Anna Trauzold for providing pancreatic cancer cell lines and Nadine Süßner for excellent technical assistance. A.L.K. and M.S. contributed equally to this work. U.K. and J.L. shared senior authorship.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
This work was funded by a research fellowship from the University of Ulm (Bausteinprogramm 3.2) given to Johannes Lemke. Work in the lab of Uwe Knippschild is funded by the Deutsche Forschungsgemeinschaft (DFG; KN356/6-1).
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.