
Abstract
Urogenital schistosomiasis is a neglected tropical disease that can lead to bladder cancer. How urogenital schistosomiasis induces carcinogenesis remains unclear, although there is evidence that the human blood fluke Schistosoma haematobium, the infectious agent of urogenital schistosomiasis, releases estradiol-like metabolites. These kind of compounds have been implicated in other cancers. Aiming for enhanced understanding of the pathogenesis of the urogenital schistosomiasis-induced bladder cancer, here we review, interpret, and discuss findings of estradiol-like metabolites detected in both the parasite and in the human urine during urogenital schistosomiasis. Moreover, we predict pathways and enzymes that are involved in the production of these metabolites emphasizing their potential effects on the dysregulation of the tumor suppressor gene p53 expression during urogenital schistosomiasis. Enhanced understanding of these potential carcinogens may not only shed light on urogenital schistosomiasis-induced neoplasia of the bladder, but would also facilitate development of interventions and biomarkers for this and other infection-associated cancers at large.
Keywords
Urogenital schistosomiasis-induced squamous cell carcinoma of the bladder
Schistosomiasis is a major neglected tropical disease (NTD) and is considered the most important human helminth disease in terms of morbidity and mortality.1,2 Control strategies have been used to block transmission and reduce the burden of this NTD, including mass targeted chemotherapy, improvements to sanitation, modification of the environment, and use of molluscides.2,3 Although different approaches to control, prevent, or in some cases even eliminate NTDs exist, like water purification or mass drug administration, for many of them, the tools and implementation strategies available are suboptimal, incomplete, or inadequate to sustain elimination effectors.1,4 Chemotherapy against flatworm infections, such as schistosomiasis and opisthorchiasis, is constrained by the limited number of indicated drugs in the market, generally only praziquantel. Emerging drug resistance has been already reported. 5 Yet schistosomiasis remains a major public health problem especially in endemic rural regions of sub-Saharan Africa. 6 Eggs are shed continuously by the Schistosoma haematobium adult parasites; these eggs engender chronic, granulomatous inflammation in the bladder wall, leading to severe disease including malignancy (Figure 1). 7 Indeed, bladder cancer is a frequent and grim complication of chronic urogenital schistosomiasis (UGS). 8 The World Health Organization’s International Agency of Research Cancer (WHO IARC) categorizes infection with S. haematobium as a definitive etiology of cancer, that is, Group 1 carcinogen.1,9 The severity and frequency of squamous cell carcinoma (SCC) of the bladder are related to the burden and duration of the infection.8,10 Understanding the mechanisms underlying the S. haematobium infection-related carcinogenesis is a key element to define targets for novel control tools-vaccines or new drugs to combat the infection and its consequences. 11

(a) Representative case of urogenital schistosomiasis (UGS)-induced bladder cancer, in a patient residing in an endemic region of UGS in Northern Angola. Arrow indicates the affected area, (b) representative micrographs of eggs of S. haematobium, both viable (A) and calcified (B) and (c) Histology of bladder mucosa revealing eggs of S. haematobium (asterisks).
Estrogen-DNA adduct-mediated pathways may underlie, at least partly, the etiopathogenesis of the UGS-induced bladder cancer.12–16 Interactions between the estrogen receptor (ER) and total antigen preparation of S. haematobium have been described in vitro. Chinese Hamster Ovary (CHO) cells transfected with a construct expressing luciferase driven by the estrogen response element (ERE) were exposed to S. haematobium adult worm antigen preparation. Luciferase activity measured in cells exposed to S. haematobium antigens was significantly lower than controls, suggesting that the antigen preparation downregulated and repressed the ER activity at least in vitro. 13 Downregulation of ER-alpha and ER-beta may serve to control physiological responses in target tissues. 17 These findings suggest that total antigens dampen the stimulatory response to estradiol. 12 In addition, schistosome may synthesize estrogen-like metabolites, and derived estrogen-DNA adducts that circulate in the blood may be excreted in the urine during UGS.12,13 The causes and consequences of the formation of these estrogen-derived metabolites, and whether they might promote DNA damage in the bladder urothelium and/or other host tissues remain unclear. Estradiol-like metabolites and novel metabolites of 8-oxo-2′-deoxiguanosine (8-oxodG) were detected in urine of UGS and associated SCC of the bladder cases; 15 8-oxodG liberated following oxidation of deoxynucleotides, has been implicated in the initiation and/or promotion of inflammation-mediated carcinogenesis, and is indicative of chromosomal damage.15,18,19 Trapped eggs in bladder tissue induce a distinct immune-mediated granulomatous response that causes local and systemic pathological effects that along with resulting bacterial superinfection and renal dysfunction also can have lethal consequences. 2 Mutations and/or expression dysregulation of p53 gene have been associated with benign and premalignant lesions and histologically normal mucosa adjacent to the SSC tissue, during UGS-induced carcinogenesis. 20 Determining whether these mutations in p53 are related to the presence of estrogen-DNA adducts might be instructive. 20
The tumor suppressor gene p53, located on chromosome 17p13, is a frequent target for mutations commonly involved in tumor progression in diverse neoplasias.21,22 Diverse roles for p53 have been described, including cell cycle control, DNA repair, activation of apoptosis, 23 inhibition of tumor growth, suppression of cell transformation, 24 and maintenance of genome integrity.25–27 Loss or mutation of p53 accelerates tumorigenesis and alters the cell response to agents that damage the DNA. 22 In particular, several mutations in p53 have been associated with UGS-induced bladder cancer, for example, mutations in the coding region, codon 249, were identified in dysplasic and metaplasic lesions in the urinary bladder or frequently in invasive SCC of the bladder.19,21,28,29
Here, we propose a pathway for the formation of these estrogen-like metabolites and describe key enzymes based on recent findings.12–16 Emphasis is given to the tentative association between these metabolites, dysregulation of p53 and DNA damage in the target tissue—the urothelium of the urinary bladder-during UGS. Estradiol-like, catechol-like, and/or oxysterol-like metabolites play a role in carcinogenesis as potential initiators of chemical carcinogenesis,15,30 and several mechanisms may explain their role in the disease. The hormone-mediated mechanism by which the ER is stimulated by some of these metabolites promoting cellular proliferation, increasing the likelihood of errors in DNA replication, has been well studied. Alternatively, estrogen metabolites acting as chemical carcinogens might react covalently with the DNA by redox cycling or by forming an abasic site. Subsequent error-prone repair of the damaged DNA eventually results in chromosomal lesions that initiate carcinogenesis.14,30–33
Pathways for formation of estrogen-like metabolites
Novel, estrogen-derived compounds, recently reported,12–15 are based on the steroid core cyclopentanophenanthrene-4-ring, that is, similar to cholesterol, a precursor of bile acids, and steroid hormones including estradiol. 34 It is feasible that S. haematobium produces estradiol-related compounds, given that this parasitic flatworm may have evolved orthologous genes to those in mammals involved in estrogen metabolism. 35 Estradiol-like metabolites of schistosome origin may underlie the development of SCC of the bladder during UGS. 36
Mammalian and related hormones, estrogen-like metabolites, and schistosome physiology
Whereas mammalian host hormones are required for the survival, development, and differentiation of schistosomes, the underlying molecular mechanisms remain unclear.36,37 The Asian schistosome, Schistosoma japonicum, expresses receptors for steroid hormones displaying pathways for processing steroid hormones to anabolize derivatives. 38 The schistosome genome encodes putative enzymes that could convert progesterone and pregnenolone to estriol, estrone androsterone, and testosterone. Hence, schistosomes might exploit this pathway during their parasitic developmental stages. 38 A gene encoding an ortholog of 17β-hydroxysteroid dehydrogenase has been identified in the genome of S. haematobium. Orthologs of 17β dehydrogenase also occur in S. mansoni and S. japonicum, but given that the infection with these species of schistosomes has not been associated with cancer development, unlike the chronic infection with S. haematobium, 36 among other factors, it is not unreasonable to speculate that specific host tissues exposed to S. haematobium eggs, that is, urothelium, might be more susceptible to DNA damaged induced by these metabolites than host tissues exposed to S. mansoni and S. japonicum eggs, that is, liver and intestine. 35 Moreover, the genomes of these three schistosomes likely encode orthologous enzymes involved on steroid biosynthesis and steroid hormone biosynthesis pathways (Supplementary Figures S1 and S2). Characterization and functional analysis of these enzymes, summarized in Tables 1 and 2, employing functional genomic tools such as transgenesis, gene silencing, and CRISPR-Cas9-mediated genome editing might provide in the near future insights in their biological roles in the host-parasite relationship including in UGS-induced SCC.39–41
Orthologs of enzymes of the steroid biosynthesis pathway encoded in the genomes of Schistosoma haematobium and S. mansoni.

Orthologs of enzymes of the steroid hormone biosynthesis pathway orthologous enzymes encoded in the genomes of Schistosoma haematobium and S. mansoni.

Role of P450 and reactive oxygen species (ROS) in the synthesis of metabolites
Steroid-related molecules may be metabolized into catechol-like compounds (intermediately producing ROS) that react with DNA via Michael addition forming DNA adducts and creating apurinic sites that could eventually lead to mutations further initiating a carcinogenesis process. Diverse cytochrome P450 enzymes play a key role both in the formation of estrogen and its subsequent oxidative metabolism as well in cholesterol catabolism to bile acids, 42 and also as a source of ROS. 43 The oxygenated metabolites of estrogens represent structures with newly generated hydroxyl and keto functions at specific sites in the steroid nucleus, which are analogous to other steroid categories that undergo oxidative metabolism, specifically androgens, vitamin D, and bile acids. 44 As shown in Figure 2, the conversion of catechol semiquinones to quinones, performed by these types of enzymes, can also enter into redox cycling and, thereby release ROS. 45 Accordingly, since S. haematobium exhibits P450 activity 28 suggesting that it is able to produce catechol compounds that lead to formation of ROS, this may contribute to oxidative stress during UGS. Increased oxidative stress associated with schistosomes has been described. 14 When ROS accumulate and/or when antioxidants defenses are saturated, the negative effects of oxidants ensue. 46 ROS induce the oxidation of DNA bases to yield 8-oxodG. 18 Thus, P450 through oxidative metabolism could lead to both oxidative and DNA adducts, with mutagenic potential. 31 The carcinogenic process in the bladders of the mice exposed to parasite antigens is independent of the effect of nitrosamines, findings indicating that schistosome antigens/metabolites induce inflammation. 47

Hypothetical pathway for formation of molecules based on estrogen metabolism mediated by P450 enzymes in Schistosoma haematobium and in sera of persons with urogenital schistosomiasis. The initial step is oxidation of estrone (E1) to estradiol (E2) performed by 17β-HSD. E1 and E2 are metabolized to 2-hydroxy forms [2-OHE1(E2)], and then oxidized to catechol quinones [E1(E2)-2,3-Q]. P450 enzymes catalyze the conversion of catechol semiquinones [E1(E2)-2,3-Q] to quinones, which may release reactive oxygen species (ROS). Catechol quinones are reactive electrophiles capable of reaction with DNA bases, including adenine (Ade) and deoxyadenosine (dA) to form DNA adducts with structures compatible with m/z values established here (m/z 624, 738, 851, 963).
Estrogen-like metabolites and DNA adducts
There is a stepwise series in biosynthesis of estrogen from cholesterol. In brief, the biosynthesis commences with formation of pregnenolone catalyzed by CYP11A. This is succeeded by the formation of the androgens androstenedione and testosterone, catalyzed by CYP17, and is completed when CYP19 catalyzes aromatization of androstenedione to yield estrone (E1) and estradiol (E2). 48 Estradiol (E2) is mainly metabolized by (i) oxidation of hydroxyl function at the C-17 position to yield estrone (E1). 17β-Hydroxysteroid dehydrogenase (17β-HSD), a key enzyme in the metabolism of estrogen, 49 catalyzes this conversion. Estrogen is also metabolized by hydroxylation via cytochrome P450 enzymes, preferentially on C2, C4, and C16 positions. When hydroxylation takes place on the steroid aromatic ring A, catechol estrogens are produced.50–52 CYP1A1, CYP1A2, and CYP3A4 participate in this process; they exhibit catalytic activity dominantly for the 2-hydroxylation in estrogen 38 to liberate 2-hydroxyestrone/estradiol (2-OHE1(E2)). Thereafter, further oxidation to semiquinones (E1E2-2,3-SQ) proceeds, and to quinones (E1E2-2,3-Q) catalyzed by P450 enzymes. ROS also are liberated in this conversion. As electrophiles, catechol quinones form covalent adducts with DNA bases, such as adenine (Ade), and deoxyadenosine (dA), via Michael addition,29,31,52,53 giving rise to structures compatible with those with m/z values 624, 738, 851, and 963 that have been identified in the peripheral blood during UGS (Figure 2). 13
Cholesterol is converted into bile acids through discrete enzymatic steps that include initiation of synthesis by 7α-hydroxylation of sterol precursors, ring structure modification, oxidation and shortening of the side chain, and conjugation of bile acid with amino acid.54,55 Microsomal cytochrome P450 enzyme cholesterol 7α-hydroxylase (CYP7A1) catalyzes the hydroxylation to yield 7α-hydroxycholesterol. 54 The product released from this hydroxylation is converted into 3-oxo Δ 4 forms by microsomal 3β-hydroxy-Δ 5 -C27-steroidoxidoreductase (HSD3B7) and forms 4-cholesten-7α-ol-3-one. 54 The enzyme CYP8B1, a sterol 12α-hydroxylase, promotes the 12-hydroxylation leading to formation of 4-cholesten-7α-12α-diol-one.54,55 Thereafter, AKR (aldo-keto reductase) family enzymes modify the aromatic ring. AKR1D1 and AKR1C4 act in concert, where AKR1D1 catalyzes 5β-reduction of the bile precursor from 4-cholesten-7α-12α-diol-3-one to 5β-cholesten-7α-12α-diol-3-one; AKR1C4 catalyzes the further reduction of ketogroups 56 forming 5β-cholesten-3α-7α-12α-triol. The next step is the side chain hydroxylation leading to form 5β-cholesten-3α-7α-12α-27-triol performed by CYP27A1. CYP27A1 introduces a hydroxyl group at C27 position and oxidizes this group to an aldehyde and subsequently to a carboxylic acid55,57,58 leading to 3α-7α-12α-trihydroxy-5β-cholestanoic acid. Bile acid CoA ligase, CoA racemase, AcylCoA oxidase, D-bifunctional enzyme, and peroxisomal thiolase 2 modify the product to 3α-7α-12α-trihydroxy-5β-cholan-24-one-CoA.55,56 The terminal step of primary bile acids formation is the conjugation with an amino acid, usually glycine or taurine, and occurs in amide linkage on C24. Bile acid coenzyme A: amino-N-acyltransferase catalyzes the reaction. The latter is a notably efficient enzyme, given that >98% of bile acids excreted from liver are amidated. 59 The substrates are a bile acid coenzyme A thioesther (3α-7α-12α-trihydroxy-5β-cholan-24-one-CoA) and glycine and taurine. 59 The ultimate step liberates glycocholic or taurocholic acid.
As obligate parasites, the blood flukes likely evolved the capacity to exploit host hormones for their own needs, deploying the enzyme P450 to release E1(E2)-2,3-Q and related metabolites including catechols and other electrophiles (Figure 2). These metabolites are generally inactivated by conjugation reactions including methylation, glucuronidation, and/or sulfation. The common pathway of conjugation in extrahepatic tissues occurs by O-methylation catalyzed by the ubiquitous catechol-O-methyltransferase (COMT). 53 If conjugation of catechol estrogens via methylation becomes insufficient, competitive catalytic oxidation of catechol estrogen to catechol estrogen quinones can proceed. 53 Alterations in the activity of phase I and phase II drug-metabolizing enzymes in the urothelium of the bladder during UGS might arise, interfering with the detoxification of xenobiotics and other homeostatic processes. Indeed, diminished glutathione-S-transferase activity occurs during UGS. 46 If homeostatic detoxification of catechols was impeded during UGS, these metabolites might accumulate in the urothelium and elsewhere.
Pioneering reports indicated that schistosomes could not synthesize sterols de novo. 60 However, this is controversial given that lysates of schistosomes convert steroids to related metabolites.42,61 Following infection of the skin penetration, the schistosomules migrate in the circulation to the lungs, after which the parasites enter the hepatic portal circulation or the vessels of the pelvic organs depending on the species; S. haematobium adult worms dwell in the venous plexus of the bladder and genital tract, whereas the other major human species dwell in the mesenteric veins. 62
Catechol-like metabolites produced either by non enzymatic process (autoxidation by oxidative free radicals) or enzymatic catalysis 42 might react with DNA bases to yield the molecules we have proposed (Figure 3). Based on recent findings,9,10,12,16 glycocholic acid may (1) lose the acidic moiety (-CH2COOH) and undergo hydroxylation then oxidation to produce the catechol-like compound m/z 410; (2) lose oxo groups, and introduce a double bond on aromatic ring A to form the compound with m/z 366; (3) forms DNA adducts by reacting with DNA bases via Michael addition, m/z 716; (4) or after aromatic ring modifications and further reaction with DNA bases to form m/z 803; or (5) undergo through a hydroxylation at C2 position followed through an oxidation and then react with DNA forming m/z 813. This compound could undergo through a reduction of some hydroxyls group forming the compound with m/z 817 (Figure 3(a)). Other compound proposed (Figure 3(b)) differs in some ways from the above. Instead of a glycocholic acid, the precursor is a taurochenodeoxycholic acid. The synthesis is similar to that for glycocholic acid, but this compound does not proceed through hydroxylation of CYP8B1. Rather than forming 5β-cholesten-7α-12α-diol-3-one, 5β-cholesten-7α-ol-3-one is liberated (Figure 3(b)). This product undergoes the same reactions of 5β-cholesten-7α-12α-diol-3-one, but the reaction conjugates the amino acid taurine instead of glycine. The next step is the reduction of sulfonic acid moiety (-SO3H) of taurochenodeoxycholic acid to a thiol group (-SH). Again, this product could eventually react with DNA bases leading to the compound with m/z 791. The estrogen-like DNA adducts with m/z 716, 803, 813, 817 have been described in S. haematobium; 14 the remaining compounds highlighted here, m/z 366, 410, 791, have been seen in urine during UGS including cases with or without bladder cancer. In addition, the m/z 803 metabolite was identified in both eggs and other developmental stages of the schistosome, and in urine of patients with UGS.14,15

Possible pathway formation for estradiol-like metabolites reported in eggs and adult stages of Schistosoma haematobium and in urine of patients with urogenital schistosomiasis (UGS), derived from glycocholic acid (a) and taurochenodeoxycholic acid (b). The molecules with m/z 366, 410, 791 were observed in urine during UGS with or without bladder cancer. Metabolites with m/z 813 and 817 were observed in schistosomes. The compound with m/z 803 was observed in urine during UGS and also within the parasite itself.
Consequences of estrogen-like metabolites
The formation of DNA adducts induces continuous DNA oxidation, leading to mutations and/or gene expression dysregulation of oncogenes and tumor suppressors, such as the overexpression of p53 observed during UGS. 63 Consequently, these metabolites of estrogens derived from the parasite might behave as chemical carcinogens. 47 In addition, there may be “classical” hormone-like effects of these metabolites on the endocrine and immune system of the host; the ER activity was suppressed in urothelial cells cultured in vitro and in the bladders of mice exposed to S. haematobium antigens. 64
The levels of oxidized DNA bases reflect a balance between the amount of oxidative DNA damage and DNA repair. It seems reasonable to assume that a suboptimal repair of 8-oxodG would be mutagenic, and may be implicated in the initiation and/or promotion of inflammation-mediated carcinogenesis.47,65,66 During UGS, in the face of continuous shedding of parasite eggs into the wall of the bladder and egress of the eggs with the urine, sustained production of catechols is expected, with continuous generation of ROS that oxidize DNA bases of urothelial cell chromosomes. Cavalieri and coworkers postulate that the presence of high levels of oxidized DNA suggests a carcinogenic mechanism induced by catechol estrogens. 52 Among the compounds related to DNA adducts, we observed metabolites with m/z 285, 274, 265, 204, 190 (Figure 4), derived directly of 8-oxodG in patients with UGS and UGS-induced bladder cancer. 15 Higher levels of metabolites derived from 8-oxodG were identified in persons with UGS free of cancer than in patients with already advanced UGS-induced bladder cancer. Hence, we speculate that DNA oxidation may be more pronounced during incipient cancer than where the UGS-induced bladder cancer has already manifested. 15 In addition, 8-nitroguanine forms via inducible expression of nitric oxide synthase in Oct3/4-positive stem cells in UGS-associated bladder cancer tissue, 67 and DNA nitrative and oxidative mutations characterized by 8-nitroguanine and 8-hydroxy-2′-deoxyguanosine (8-oxodG), have been implicated in the promotion of inflammation-mediated carcinogenesis by infection with S. haematobium. 68 Moreover, other studies have shown that UGS is likely to cause bladder cancer by this mechanism, and noted a correlation between UGS and increase of levels of oxidative stress accompanied by continuous DNA damage and repair in urothelial carcinomas.20,69 These metabolites might damage DNA, for example, by formation of DNA adducts (m/z 624, 716, 738, 803, 817, 851, and 963) and contribute to intermediate production of ROS during formation of catechol compounds (Figures 2 and 3). Furthermore, these phenomena may explain formation of 8-oxodG derivatives (as illustrated in Figure 4), metabolites known from urine during UGS. 15

DNA damage possibly related to estrogen-like metabolites from parasite. Compounds in urine of persons infected with the blood fluke S. haematobium with or without infection-induced bladder cancer. The compounds derive directly from 8-oxodG, a product of DNA oxidation. Formation of these molecules may be a consequence of interaction with catechol compounds from parasite.
Overexpression of p53 during UGS
The p53 is a tumor suppressor protein involved in diverse pathways including regulation of cell cycle, suppression of cell proliferation, cellular response to DNA damage, initiation of DNA repair and replication, induction of apoptosis, and promotion of differentiation.27,28,66,70 Dysregulation of the expression of p53 gene may accelerate the tumorigenesis and alter the response of cellular agents that damage DNA. 2 Together, the genes encoding these proteins are referred to as tumor suppressor genes. Inactivation or alteration in the expression of these genes together with activation of oncogenes leads to malignant transformation. 71 It has been shown that the formation of DNA adducts may be related to a high expression of p53 during schistosomiasis.28,72,73 Given that (i) S. haematobium produces compounds that react with DNA and damage chromosomal DNA, and (ii) p53 mutations have been associated with schistosomiasis, 28 chronic infection with S. haematobium may stimulate the expression of p53 during UGS and UGS-induced bladder cancer. In addition, p53 is overexpressed during UGS, although the molecular mechanisms remain unclear,20,23,29,69,74 and p53 protein has been detected by immunolocalization in both aggressive urothelial and squamous cell bladder carcinomas. Notably, p53 is mutated and not functional in benign/premalignant lesions, predominantly in those showing cellular alterations, including urothelial hyperplasia, epidermoid metaplasia, and/or dysplasia. 20 This conforms to findings in urine using liquid chromatography–mass spectrometry LC-MS/MS during UGS, where DNA oxidation processes appear more pronounced in cases with UGS but without cancer. 15 The eventual association between the dysregulation of p53 in urothelium and infection with S. haematobium reinforces the notion that UGS profoundly alters in cellular process in the urothelium, ultimately giving rise to bladder cancer after a continuous accumulation of mutations and selection of aggressive clones of transformed cells. By employing a model of chemical-induced carcinogenesis of the bladder in genetically modified mice, that is, using N-butyl-N-(4-hydroxybutyl)nitrosamine (BBN), it has been recently demonstrated the cellular origin of the bladder neoplasia. 75 In this model, invasive carcinoma is initiated from basal urothelial stem cells that started to accumulate mutations that eventually offer positive selection of aggressive clones of cells. The probability of this phenomenon ultimately depends on the mutagenesis pressure on the target tissue that in the context of UGS where the described estrogen-like metabolites are produced may be higher. 15
Future perspectives
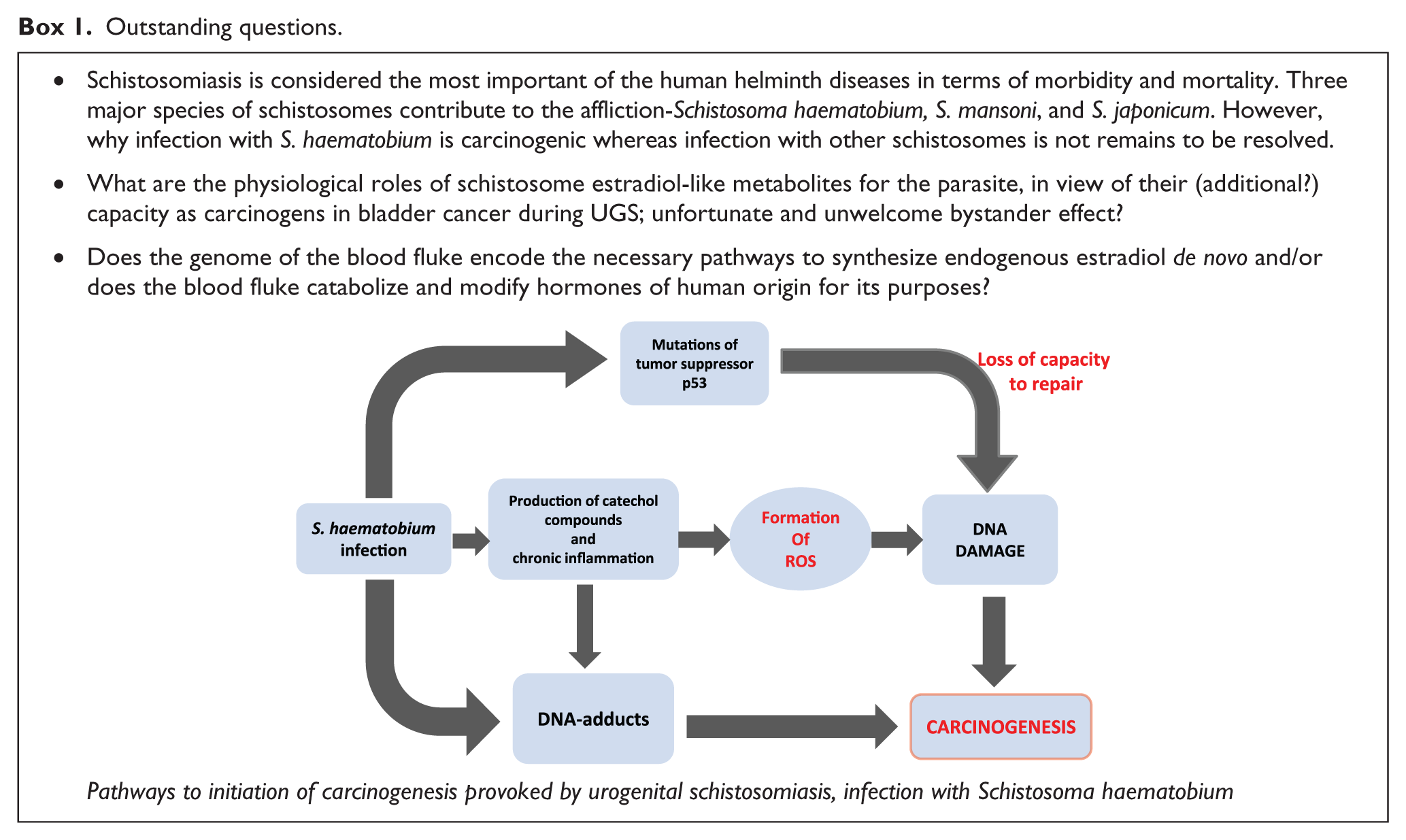
Carcinogenic transformation of the bladder during UGS may proceed along a pathway involving the formation of catechol and DNA adducts, intermediary formation of ROS, p53 mutations that inhibit the repair of DNA and, in turn, initiate carcinogenesis (Box 1). Whereas carcinogenesis may be induced by other factors,76,77 this is a plausible route to UGS-induced SCC. Pathways for formation of these carcinogenic metabolites with parasite origin are not yet fully understood. Investigations in this field are required to determine specific enzymatic functions of pathogens and whether these correlate with the ability of various Schistosoma species to produce bladder cancer. The findings hold the promise for new interventions, including prognostic biomarkers and indeed a vaccine against this NTD-induced cancer.
Outstanding questions.
Footnotes
Declaration of conflicting interests
N.V. thanks Fundação para a Ciência e Tecnologia (FCT, Portugal) and FEDER (European Union) for funding through UID/MULTI/04378/2013, project grant IF/00092/2014, and IF2014 position. Thanks are also due to Comissão de Coordenação e Desenvolvimento Regional do Norte (CCDR-N)/NORTE2020/Portugal 2020 for funding through project DESignBIOtechHealth (ref. Norte-01-0145-FEDER-000024). J.M.C.C. thanks FCT for funding through UID/Multi/00211/2013 (01.01.2015—31.12.2016—Centro de Estudos de Ciência Animal). P.J.B. and G.R. received support from awards R01CA155297 and R01CA164719 from the National Cancer Institute (NCI), National Institutes of Health (NIH). The content is solely the responsibility of the authors and does not necessarily represent the official views of the FCT, NCI, or NIH.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.