
Abstract
Prostate-derived Ets factor (PDEF), a member of the Ets family of transcription factors, differs from other family members in its restricted expression in normal tissues and its unique DNA-binding motif. These interesting attributes coupled with its aberrant expression in cancer have rendered PDEF a focus of increasing interest by tumor biologists. This review provides a current understanding of the characteristics of PDEF expression and its role in breast cancer. The bulk of the evidence is consistent with PDEF overexpression in most breast tumors and an oncogenic role for this transcription factor in breast cancer. In addition, high PDEF expression in estrogen receptor–positive breast tumors showed significant correlation with poor overall survival in several independent cohorts of breast cancer patients. Together, these findings demonstrate PDEF to be an oncogenic driver of breast cancer and a biomarker of poor prognosis in this cancer. Based on this understanding and the limited expression of PDEF in normal human tissues, the development of PDEF–based therapeutics for prevention and treatment of breast cancer is also discussed.
Keywords
Introduction
Prostate-derived Ets factor (PDEF), also known as SPDEF (Sam-pointed domain containing Ets transcription factor), is a member of the Ets family of transcription factors. This transcription factor family is evolutionarily conserved in metazoan with 10 and eight members, respectively, in Caenorhabditis elegans (C. elegans) and Drosophila and a much larger number, that is, 27 and 26 Ets genes, respectively, in humans and mice. A characteristic feature of the Ets genes is an 85 amino acid long DNA-binding domain consisting of a winged helix turn helix structure that binds to DNA at a conserved GGAA/T sequence motif. Phosphorylation by Ras–MAP (mitogen-activated protein) kinase pathway is an important mode of activation of these Ets transcription factors. They play an important role in a number of cellular processes including regulation of cell differentiation, proliferation, angiogenesis, and apoptosis. Moreover, they are aberrantly expressed in specific leukemias and solid tumors. A number of excellent reviews have previously discussed these features of the Ets transcription factor family in normal development and in cancer.1–6
PDEF was originally described as a prostate-specific molecule in both humans and mice.7,8 Its protein sequence consists of an N-terminal transactivating domain containing specific phosphorylation sites as putative substrates for Ras–MAP kinase and other signaling pathways; a protein–protein interacting Sam-pointed domain; and a small linker segment of unknown function. This is followed by the C-terminal DNA-binding domain. 7 In contrast to other members of the Ets family that bind to the GGAA sequence on DNA, PDEF preferentially binds to the GGAT sequence for its transcription activity.7,9 Furthermore, in the LNCaP prostate tumor cell line, PDEF was shown to bind to the prostate-specific antigen (PSA) promoter sequence, alone or in interaction with an androgen receptor (AR), to enhance transcription from the PSA promoter. 7 PDEF-mediated transcription from the PSA promoter was later shown to be inhibited by NKX3.1, a well-known tumor suppressor in prostate cancer. 10 Moreover, PDEF expression itself was found to be induced by androgen signaling in LNCaP prostate tumor cell line. 11 Together, these findings suggested an important function for PDEF in normal prostate development and a role in prostate cancer progression.7,10,11 However, over the years, contradictory results were published that have created divergent views about the role of PDEF in cancer. We previously addressed the issue of PDEF’s role in prostate cancer. 12 The purpose of this review is to provide a current understanding of the characteristics of PDEF expression and its role in breast cancer. The bulk of the evidence is consistent with PDEF overexpression in most breast tumors and an oncogenic role for this transcription factor in breast tumor progression. Other specific observations that suggest an apparent tumor suppressor role for PDEF in breast cancer are also discussed.
Overexpression of PDEF in breast cancer
A number of studies have reported overexpression of PDEF messenger RNA (mRNA)13–16 and protein 11 in breast tumors compared with benign breast tissues. In our study, we used a validated anti-PDEF antibody raised against an N-terminal 104 amino acid residue long fragment unique to PDEF, 11 to compare PDEF protein expression in matched benign and tumor samples from nine breast cancer patients. In eight of these cases, PDEF was significantly overexpressed in the tumor tissue compared with the adjacent benign tissue. Representative photomicrographs from three matched cases are shown in Figure 1. These data demonstrated PDEF protein overexpression in most breast tumors. Another study, on the contrary, reported loss of PDEF protein expression in the tumor tissue compared with normal breast tissue. 17 However, the specificity of the antibody for PDEF was not demonstrated in an immunohistochemistry (IHC) assay in that study. The latter validation is critical in view of the knowledge that many Ets genes are expressed in individual normal human tissues 18 and likely also in breast cancer. This raised the potential for cross reaction of PDEF antibodies with other Ets proteins and perhaps also with other unknown proteins.

Staining of matched pairs of benign and tumor tissue samples from breast cancer patients with anti-PDEF antibody: As shown in this figure, the tumor tissues have stronger nuclear staining of epithelial cells. In addition, much higher fractions of the epithelial tumor cells stained positive for PDEF compared with epithelial cells in benign breast tissues.
IHC validation of antibody specificity is also necessary since PDEF epitopes detected in the IHC assay may be different from those seen on western blotting. This validation may be achieved by testing the reactivity of a PDEF antibody on a panel of normal human tissues with known PDEF mRNA expression characteristics. When such rigorously IHC validated anti-PDEF antibodies were used to analyze PDEF expression in benign breast and breast tumor samples, overexpression of PDEF was observed in the tumor samples compared with benign tissues. 11 These results were later supported by our analysis of 131 primary breast tumors that showed 10-fold or higher expression of PDEF mRNA in 72% of these tumors. 15
Similar to human breast tumors, overexpression of prostate-specific Ets (PSE; the mouse homolog of PDEF) was also reported in the spontaneously arising mammary tumors from rat neu transgenic mice and from polyomavirus middle T antigen (PyMT) transgenic mice. 19 Together, these results demonstrate widespread PDEF overexpression in breast tumors from independent cohorts of breast cancer patients and in spontaneously arising mammary carcinomas of mice.
PDEF, an oncogenic driver of breast cancer
Transfection of PDEF increased the colony-forming, migratory potential and tumorigenicity of the pre-malignant MCF-10A and MCF-12A breast epithelial cell lines in vitro and in immunodeficient mice, respectively.20,21 Conversely, PDEF downregulation by small interfering RNA (siRNA) led to the loss of the viability of malignant breast tumor cell lines in vitro and growth in vivo.15,16 Remarkably, a genome-wide shRNA screen using 77 human breast tumor cell lines independently identified FOXA1 and SPDEF (another name for PDEF) as the two most essential genes for the survival/growth of luminal (ER+, Her2+) breast tumor cell lines. 22 Together, these observations showed that PDEF is a highly active oncogene in breast cancer. We recognize that other studies have suggested PDEF to be a tumor suppressor.17,23,24 However, a specific pitfall of these studies was that highly malignant basal subtype breast tumor cell lines were used in PDEF transfection experiments. Similar transfection of the estrogen receptor (ER) complementary DNA (cDNA) into the malignant ER-negative MDA-MB231 breast tumor cell line also showed reduced growth of these cells in vitro following treatment with estradiol. 25 These results are contrary to the well-established paradigm that estrogen signaling through ER is oncogenic since inhibiting this signaling by tamoxifen results in about 50% reduction in the incidence of ER+ breast cancer. These observations suggest that already malignant basal subtype breast tumor cell lines such as MDA-MB231 are not useful models to study the role of a putative cancer gene in tumor progression. Presumably, malignancy of these cell lines may not be susceptible to further increase following transfection with PDEF or ER. Rather, these luminal epithelial specific transcription factors may reduce the mesenchymal phenotype of these cell lines, rendering them less invasive and less migratory.17,23 Indeed, classically, pre-malignant cell lines such as NIH 3T3 cells were used in transfection experiments to determine the transforming capacity of putative cancer genes. Similarly, the individual genes described in the review article 24 to ascribe a potential tumor suppressor role for PDEF were mostly derived from the studies that transfected PDEF into malignant tumor cell lines. Hence, their proposed relevance to the role of PDEF as a tumor suppressor in breast cancer should be viewed with caution.
Further support for the notion that PDEF is not a tumor suppressor may be derived from the following considerations. Typically, a tumor suppressor is frequently inactivated in cancer. In contrast, few inactivating mutations in the PDEF gene were described in the hundreds of breast tumors that have been sequenced to date. 26 Rather, PDEF is overexpressed in most primary human breast tumors11,13–16 (Figure 1), as is its mouse homolog in the spontaneously arising mouse mammary tumors. 19 In addition, the fibroblast growth factor receptor 2 (FGFR2) locus polymorphism associated with increased breast cancer risk is characterized by the increased transcriptional activity of SPDEF. 27 Clearly, the vast majority of the evidence is consistent with an oncogenic role of PDEF in breast cancer.
Strong evidence for a pro-survival role of PDEF in breast cancer was also derived from the analysis of genes regulated by PDEF in the MCF-7 breast tumor cell line. 16 Gene ontology analysis of differentially regulated genes in PDEF-knockdown and control MCF-7 cells showed downregulation of cell cycle–related genes and upregulation of apoptosis-related genes in PDEF-knockdown cells. Notably, PDEF was shown to downregulate FAS receptor expression, thereby inhibiting the apoptosis of tumor cells. 16 We have also reported that carcinoembryonic antigen–related cell adhesion molecule 6 (CEACAM6) is a PDEF-induced gene in MCF-7 cells, and demonstrated elevated co-expression of these molecules in 72% of primary human breast tumors. 15 Moreover, chromatin immunoprecipitation with an anti-PDEF antibody showed enrichment for CEACAM6 gene sequences, confirming that the CEACAM6 gene is a direct downstream target of PDEF transcriptional activity (Mukhopadhyay A and Sood AK, our unpublished data). In addition, similar to PDEF, downregulation of CEACAM6 by siRNA led to the loss of viability of BT-474 and SKBR3 breast tumor cell lines in vitro. 15 Based on these results and other published work with colon and pancreatic tumor cell lines,28,29 elevated CEACAM6 expression evidently contributes to enhanced tumor cell survival by inhibiting anoikis (apoptosis caused by loss of anchor). These results suggest that inhibition of apoptosis and anoikis by PDEF-regulated genes should provide strong survival signals to PDEF-overexpressing tumor cells. Further analysis suggested that PDEF may promote tumor cell growth by inducing specific cell cycle–related genes to maintain a proliferative state. 16 These findings are again consistent with an oncogenic role for PDEF in breast cancer.
To determine whether elevated PDEF expression also occurs in high-risk pre-malignant disease, PDEF expression was analyzed in atypical ductal hyperplasia (ADH) 20 and ductal carcinoma in situ (DCIS) lesions 11 and found to be frequently elevated in both these lesions. DCIS comprises about 25% of mammographically detected breast cancers, and constitutes as many as 50,000 new cases annually in the United States. A subset of these lesions has the propensity to progress to invasive carcinomas. 30 Again, given the oncogenic properties of PDEF, its elevated expression in pre-malignant breast lesions suggests an early role for PDEF in the development of these lesions. To that end, it is noteworthy that elevated CEACAM6 (a PDEF-induced gene, see above) expression in ADH lesions was shown to be associated with the development of invasive breast cancer. 31 These observations support a role for PDEF in early breast tumor progression.
Published work also described an association of elevated PDEF expression in primary breast tumors with metastases to lymph nodes. 14 Furthermore, >80% of lymph node metastases showed overexpression of human PSE (another name for PDEF). 32 Circulating tumor cells from breast cancer patients were also found to express PDEF.13,33 Based on the oncogenic properties of PDEF, these findings support a continuing role for PDEF in breast cancer progression.
PDEF as a biomarker and target in breast cancer
As a transcription factor, PDEF may alter the expression of hundreds of genes. This was evident from the studies on changes in gene expression following modulation of PDEF gene expression in breast tumor cell lines.15,16 These results suggested that PDEF is likely to have a major influence on the biology of an epithelial/tumor cell and this should translate into a stable prognostic impact on tumor progression. To test this hypothesis, we and others have searched the Gene Expression Omnibus and Oncomine databases that contain gene expression profiling and clinical outcome data from several previously published studies, and looked for any correlation between PDEF expression and clinical outcome. The results showed that elevated PDEF expression in ER+ primary breast tumors correlated with poor overall survival in several independent cohorts of breast cancer patients.16,21 Since patients with ER+ breast tumors are likely to have been treated with endocrine therapy, correlation of PDEF with poor overall survival suggests that high PDEF expression in primary breast tumor may predict resistance to endocrine therapy. Indeed, increasing doses of the antiestrogens tamoxifen and fulvestrant resulted in higher toxicity to MCF-7 cells in vitro under conditions of PDEF knockdown by siRNA. 16 Further studies are needed to test this hypothesis and the potential of PDEF as a prognostic and/or predictive biomarker in breast cancer.
Targeting of transcription factors is a well-established paradigm in oncology as ER and AR, both of which are transcription factors, are important targets in breast and prostate cancer, respectively. As mentioned previously, a transcription factor can significantly impact the biology of a tumor by inducing large-scale changes in gene expression. Some of these genes may encode the cell surface and/or secreted molecules capable of influencing the behavior of the neighboring tumor and/or stromal cells. This concept is supported by the characteristics of ER expression in breast cancer and its implication for endocrine therapy. Thus, even when a small fraction (10%) of breast tumor cells express ER, that tumor is scored positive for ER expression and endocrine therapy shows clinical benefit for the patient.34,35 Presumably, the small fraction of ER-positive tumor cells may influence the biology of the neighboring ER-negative tumor and/or stromal cells such that inhibiting the ER-positive tumor cells by endocrine therapy compromises the viability of the rest of the tumor. In a similar vein, eliminating PDEF-expressing cells from breast tumors by PDEF-targeted therapeutics may not only kill PDEF-expressing tumor cells but also modulate the tumor microenvironment and may compromise the viability of most of the tumor tissue.
Published work shows highly restricted expression of PDEF in normal human tissues.7,11,13,14 Strong PDEF expression was observed in normal prostate and salivary glands. Weak expression in normal trachea/bronchus, colon, and stomach tissues was also reported.14,36 In contrast, others have reported little PDEF expression in the colon and stomach tissues,7,14 suggesting that the reported weak expression in these tissues may not be constitutive and may result from exposure to some inflammatory stimulus. Indeed, exposure to allergen house dust mites, cigarette smoke, or type 2 cytokines interleukin (IL)-4 and IL-13 induced PDEF expression in normal mouse trachea. 37 Similarly, contrary to another report, our IHC analysis of a number of samples from normal ovaries turned out to be negative for PDEF expression. 38 The overall limited expression of PDEF in normal human tissues suggests that PDEF-based anti-tumor therapies will have minimal toxicity against vital normal tissues. These observations support PDEF as a highly desirable novel target in breast cancer. In Table 1, salient features of PDEF in breast cancer are summarized.
Salient features of PDEF in breast cancer.
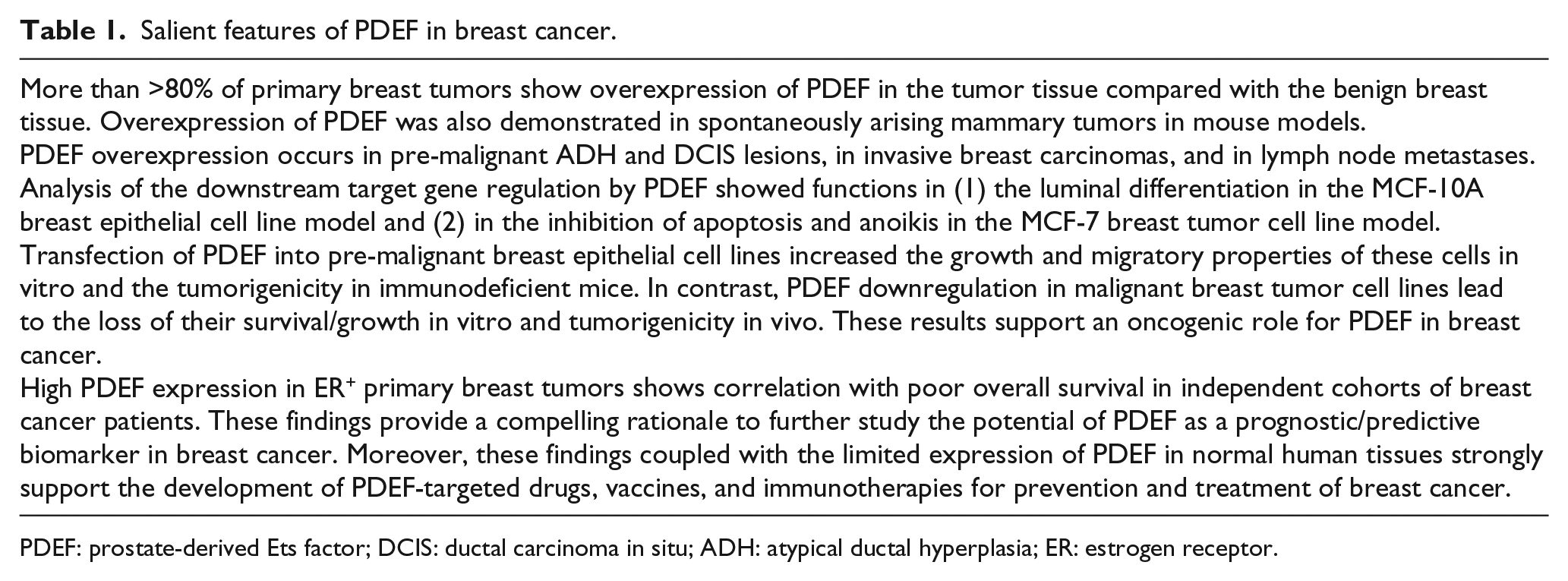
PDEF: prostate-derived Ets factor; DCIS: ductal carcinoma in situ; ADH: atypical ductal hyperplasia; ER: estrogen receptor.
Footnotes
Author’s Note
We recently analyzed PDEF expression in relation to estrogen induced molecules in the Cancer Genome Atlas (TCGA) breast cancer cohort consisting of 1095 primary breast tumors and the associated RNA-seq data. We found that PDEF expression showed significant negative correlation with the expression of estrogen induced molecules. These results are consistent with the notion that PDEF expression occurs independent of the estrogen signaling in breast tumors and that PDEF should be a useful novel target for co-targeting with endocrine therapy to minimize endocrine resistance in breast cancer (Jianmin Wang and Ashwani K Sood, our unpublished data).
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by a grant from National Cancer Institute (NCI), R41 CA84167, and a grant from the Roswell Park Alliance Foundation. The institute core facilities used in this research were supported by Roswell Park Cancer Institute and NCI grant P30CA016056.