
Abstract
The newly discovered severe acute respiratory syndrome coronavirus 2 (SARS-CoV2) has turned into a potentially fatal pandemic illness. Numerous acute kidney injury (AKI) cases have been reported, although diffuse alveolar destruction and acute respiratory failure are the major symptoms of SARS-CoV-2 infection. The AKI, often known as a sudden loss of kidney function, carries a greater risk of mortality and morbidity. AKI was the second most frequent cause of death after acute respiratory distress syndrome (ARDS) in critically ill patients with coronavirus disease 2019 (COVID-19). While most patients with COVID-19 have moderate symptoms, some have severe symptoms, such as septic shock and ARDS. Also, it has been proven that some patients have severe symptoms, such as the failure of several organs. The kidneys are often affected either directly or indirectly. The major signs of kidney involvement are proteinuria and AKI. It is hypothesized that multiple mechanisms contribute to kidney injury in COVID-19. Direct infection of podocytes and proximal tubular cells in the kidneys may lead to acute tubular necrosis and collapsing glomerulopathy. SARS-CoV2 may also trigger a cascade of immunological responses that lead to AKI, including cytokine storm (CS), macrophage activation syndrome, and Toll-like receptor type-4 activation (TLR-4). Other proposed processes of AKI include interactions between organs, endothelial failure, hypercoagulability, rhabdomyolysis, and sepsis.
Furthermore, ischemic damage to the kidney might result from the decreased oxygen supply. This article focuses on kidney injury’s epidemiology, etiology, and pathophysiological processes. Specifically, it focuses on the CS and the role of TLR-4 in this process. To effectively manage and treat acute kidney damage and AKI in COVID-19, it is crucial to understand the underlying molecular pathways and pathophysiology.
Keywords
Abbreviations
Introduction
Acute kidney injury (AKI) and kidney defects, including proteinuria and hematuria, were recently discovered as a concern during coronavirus disease 2019 (COVID-19),1, 2 which can affect up to 37% of patients and could begin concurrently with mechanical ventilation. 3 AKI is identified by a drastic drop in kidney function and is linked with an elevated risk of morbidity and death. The second most common reason for mortality in severely sick severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) patients was AKI after acute respiratory distress syndrome (ARDS). 4 The SARS-CoV2 may enter cells directly via the angiotensin-converting enzyme 2 (ACE2), where it is found in high concentrations in the kidney, causing multi-organ dysfunction. Furthermore, a viral infection may trigger inflammatory mediators and cytokine storms (CS), resulting in microvascular damage and AKI.3, 5 CS is one of the primary causes of severe COVID-19 patients’ poor prognosis, which includes multiple organ failure and mortality.6–8 The initial failure to remove the virus and, consequently, the existence of serious COVID-19 clinical symptoms have been closely associated with Toll-like receptors (TLRs). 9 The entry and replication of viruses activate different TLRs and downstream signaling pathways.10, 11 TLRs are essential in expressing and releasing various inflammatory mediators. 12 TLR especially type 4, is found on macrophages, dendritic cells, monocytes, and neutrophils. 9 Besides, it is found in many organs, including the kidney, that can recognize pathogen-associated molecular patterns (PAMPs), which are expressed by pathogens. 13 Therefore, CS should be urgently treated; Mortality will otherwise be expected to occur. 6 In addition to treating the disease, the anti-viral may specifically attack the virus. It is proposed to suggest anti-inflammatory treatments that lower cytokine responses, mortality, and morbidity. 14 Therefore, the effective suppression of CS by targeting Toll-like receptor type-4 activation (TLR-4) represents an important technique to stop the deterioration of patients with AKI complicating COVID-19. 6 TLR-4 inhibition by TLR-4 antagonist is assumed to have a key role in preventing the progression of AKI. 15
This article briefly summarizes the epidemiology, etiology, and pathophysiology pathways contributing to kidney disease in COVID-19. More precisely, it focuses on the CS and the role that TLR-4 plays in the process. In order to properly manage and treat this illness, it is vital to have a solid understanding of the underlying molecular processes and pathophysiology of acute kidney damage and AKI in COVID-19. Only then can this condition be managed and treated adequately.
COVID-19 Overview
A new virus called COVID-19 was discovered in Wuhan, China, in December 2019. The virus infection belongs to the ribonucleic acid (RNA) beta coronavirus family, identified as SARS-CoV-2. 7 In late January 2020, this virus was announced as a worldwide health epidemic by the World Health Organization (WHO), and a pandemic was announced in March 2020. In May 2020, COVID-19 reported cases reached three million worldwide, with a mortality rate of more than 250,000. 16 The recently evolving COVID-19 affects global medical health services, deteriorating the situation. 17 At the start of the pandemic, neither a vaccine nor anti-viral medications were available to treat SARS-CoV-2. Thus, travel restrictions, interpersonal distance, and patient isolation were used. 18 Based on several studies, this disease may be an either asymptomatic or mild infection or may have severe symptoms such as respiratory failure. The majority of cases, however, necessitate Intensive Care Unit (ICU) admission due to ARDS involving mechanical ventilation assistance. Some patients with severe cases of COVID-19 develop cytokine storm syndrome (CSS) and inflammatory response and one of its most important features is overwhelming systemic inflammation and fatal multi-organ failure. 19
Complication of COVID-19
Even though SARS-CoV-2 mostly affects the respiratory system, earlier research suggests that it also affects other organs and leads to organ failure and, eventually, death. 20 The development of systemic illness, defined by multisystem organ damage or failure, is one of the primary indicators of COVID-19 severity. 21 AKI is a significant COVID-19 complication.22–24 It has been shown that adverse renal symptoms, such as AKI, are associated with the severity or mortality of COVID-19. 25
Acute Kidney Injury
AKI is frequently detected in critically ill patients, with a 50% incidence rate in ICU patients. 26 AKI is caused by a drastic loss of kidney function, leading to an accumulation of waste products such as creatinine, urea, and uric acid. AKI is significantly related to an increase in morbidity and death rates. The disease is characterized by an unexpected and sudden rise in serum creatinine (SCr) levels or a drop in urine output. 27 Acute renal disease is accompanied by signs of functional or structural impairment for no more than seven days. 28 Clinical criteria for this disease depend on the Kidney Disease Improving Global Guidelines (KDIGO) 2012, which include the Acute Kidney Injury Network (AKIN) and Risk, Injury, Failure, Loss, and End-stage kidney disease (RIFLE) criteria. The following clinical circumstances might be utilized to determine AKI as a diagnosis: an elevation in creatinine serum levels ≥0.3 mg/dL in 48 h; an elevation ≥1.5 times baseline during the past seven days; or a urine volume ≤0.5 mL/kg/h for 6 h.29, 30 Moreover, KDIGO proposes that AKI should be staged according to severity. It depends on the SCr increase or drops in urine output. The first stage is characterized by a 1.5–1.9-fold rise in SCr baseline or a decrease in urine excretion of 0.5 mL/kg/h for more than 6–12 hours. Stage two is defined as a 2.0–2.9-fold rise in SCr baseline or a 0.5 ml/kg/h drop in urine output over 12 hours or more. Stage 3 is defined as a threefold increase in SCr, a reduction in urinary output to 0.3 mL/kg/h for 24 h or less or longer, or anuria lasting 12 h or more. 31
AKI and kidney replacement therapy (KRT) incidence in hospitalized COVID-19 patients has ranged from 0.5% to 40% over the original baseline.32, 33 It could begin concurrently with mechanical ventilation. 3 According to a multi-centre study by Li et al., COVID-19 patients who suffered from AKI have a 5.3-fold greater mortality rate than those who did not get AKI. According to Yang and Li’s study, AKI was the second cause of death in critically ill individuals with COVID-19 after ARDS. 4
Epidemiology of AKI in COVID-19
The clinical situation affects AKI’s reported incidence and severity of COVID-19. The KDIGO standard definition of AKI has been utilized in most research. AKI affects between 30% and 50% of COVID-19 patients who are hospitalized, with the percentage increasing among those who need intensive care.23, 34–37 Several case reports show a range of AKI rates associated with COVID-19. Researchers in China found that 5%–29% of patients diagnosed with COVID-19 had AKI within 7–14 days of hospital admission, whereas researchers in the United States found far higher rates of 37%–57% in COVID-19 patients.3,4,23,38–40 An estimated 29% of all ICU patients suffer from AKI, which rises to 78% among intubated patients. 41 According to other research, up to 20% of ICU patients need KRT.38, 42–45 During the same hospitalization period, Fisher et al. compared the outcomes of 3345 patients with COVID-19 against those of 1,265 patients without COVID-19. COVID-19-positive patients were at a greater risk of developing AKI than those who tested negative (57% vs. 37%), and a larger percentage of COVID-19-positive individuals experienced stage 3 AKI than negative patients (7.2% vs. 2.7%). 40 In multi-centre research by Li et al., those patients with SARS-CoV-2 who suffered from AKI had a death rate that was 5.3 times greater than those who did not suffer from AKI. 4 The incidence of AKI was 46% among 3,993 inpatients in a New York City healthcare system, with 39%, 19%, and 42% of those cases being KDIGO stages 1, 2, and 3, respectively. 42 Similar findings were seen in a second large cohort of New York patients, which included 3,854 persons. In this cohort, 39.9% of the patients who had COVID-19 developed AKI, with 42.7%, 21.8%, and 35.5% and their stages are 1, 2, and 3 of the disease, respectively. 46 Moreover, Yang and Li’s research showed that AKI was the second leading cause of mortality in ICU patients who were diagnosed with SARS-CoV-2, behind only ARDS. 4 Another research from Saudi Arabia by Allemailem et al. found that 23.3% of COVID-19 patients had AKI. 47 The incidence of AKI was 36% among COVID-19 hospitalized patients at a Saudi Tertiary Care Medical Center. 48 A meta-analysis conducted in Saudi Arabia found that 26% (95% CI 19%–33%) of COVID-19 participants had AKI. 49
Risk Factors for AKI Development in COVID-19
There is evidence from Chinese and American research that the risk factors for AKI in severely sick COVID-19 and ARDS patients are comparable to those previously reported for AKI in the ICU (Table 1).
Potential Risk Factors for COVID-19-associated Acute Kidney Injury.
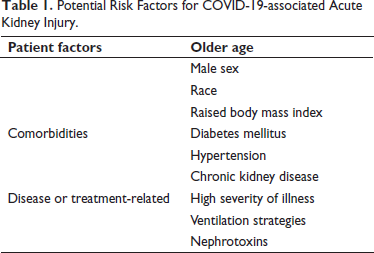
Patients with COVID-19 have been shown to have a higher risk of severe AKI when their body mass index is higher. 50 Researchers found that the male gender and older age were major risk variables for AKI. 40 Black Americans in the United States seem to have a greater relative incidence, which may be due to several variables, including genetic predisposition, socioeconomic status, and other medical conditions. 50
Patients with a severe COVID-19 course tend to have underlying health problems. The three most common comorbidities are obesity, type 2 diabetes, and high blood pressure. They are considered the most important predictors of a more severe, even life-threatening, COVID-19. AKI is prevalent in people with specific comorbidities.3, 23, 50 According to Henry and Lippi’s meta-analysis, individuals with chronic kidney disease (CKD), should avoid unnecessary SARS-CoV-2 virus infection because of the increased risk of severe disease. 51
AKI and its severity are all linked to higher levels of indicators of systemic inflammation. 38 Mechanically ventilated patients had a higher incidence of AKI and a higher requirement for KRT than non-ventilated patients in a cohort of over 5,000 Americans; the difference was statistically significant. AKI in individuals with COVID-19 may also be preceded by using nephrotoxic medications and adopting a fluid-restrictive resuscitation regimen. 3
Pathogenesis of AKI in COVID-19
Several factors are included in the pathogenesis of AKI caused by COVID-19. These factors include direct and indirect viral infection pathways, which are still poorly understood and need more investigation. The following pathogenesis will be the centre of our attention here.
Direct Attack by the Virus of SARS-CoV-2
Numerous studies have established that ACE2 is the SARS-CoV-2 host cell receptor. 52 Importantly, it must bind viral spike (S) proteins to cellular receptors. Moreover, the priming of S proteins by transmembrane serine protease 2 (TMPRSS2).53, 54 The S protein is encoded by the genome of SARS-CoV-2, and it is needed for virion assembly and infection. 55 The ACE2 receptor binding affinity is 10- to 20-fold more than SARS-CoV, which utilizes the same cellular receptor. 52 ACE2 is a type I membrane protein abundantly expressed in many human organs. ACE2 is not only present in alveolar cells of the lungs but has also been detected in the kidney and other organs, indicating that they are also susceptible to SARS-CoV-2 infection.56, 57 The intrinsic renal cells would be damaged as a result of the consequence of the virus’s binding to the ACE2 receptor. The highest concentrations of ACE2 were found in podocytes and proximal tubular epithelial cells of the kidneys.58–60 Following the respiratory tract infection, the virus reached the bloodstream and caused viremia, according to autopsy findings. It has been hypothesized that the virus enters the urinary system through the bloodstream, binds to and enters with ACE2 receptors, and infects kidney cells that express the ACE2 receptor, including renal tubular epithelial cells, podocytes, and others. As a result, the development of AKI is most likely brought on by the SARS-CoV-2 virus directly infecting kidney cells. 58
Renin-Angiotensin-Aldosterone System Impairment by SARS-CoV-2
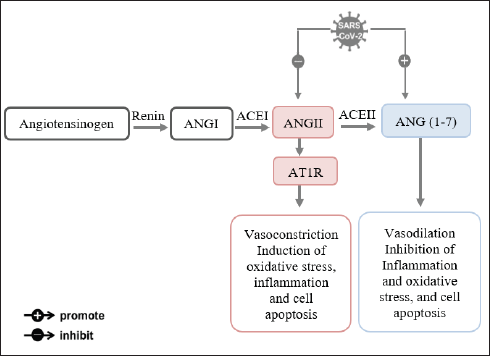
It is generally accepted that internalization of the ACE2 virus-protein complex causes a decrease in surface membrane ACE2 in COVID-19 and that this decrease makes the infected organs more susceptible to Ang II. ACE2, an ACE homologue, is a powerful anti-peptide of the renin-angiotensin system (RAS) that counteracts the effects of ACE, as it is known that the RAS is essential for blood pressure regulation and water-salt balance. Renin generates angiotensin I (ANG I) by cleaving angiotensinogen, which is then converted into angiotensin II (ANG II) by ACE cleavage. Two G protein-coupled receptors, angiotensin II receptor type 1 (AT1R) and angiotensin II receptor type 2 (AT2R), are involved in the biological effects of ANG II, including vasoconstriction, hypertension, the induction of oxidative stress, inflammation, and cell apoptosis. Through the cleavage of a terminal peptide, ACE2 breaks down ANG II to produce angiotensin (1–7) (ANG1–7). According to evidence, ACE2 exhibits protective properties such as vasodilation, the inhibition of inflammation and oxidative stress, and cell death while negatively regulating the activated RAS.61–64 ANG1−7 binds to a G-protein-coupled Mas receptor to perform its function (MasR). During infection with SARS-CoV-2, ACE2’s outer domain is cleaved and internalized after binding. So, it results in the downregulation of ACE2 and a rise in ANG II levels by lowering ANG II’s breakdown into ANG1–7. AKI worsens due to the increased oxidative stress response, cytokine production, disruption of the glomerular filtration barrier, and tissue damage brought on by ACE2 depletion, which encourages the activation of AT1R by AT2R (Figure 1).65–68
Overactivation of TLR-4
Pattern recognition receptors (PRRs) play a critical role in protecting hosts against microbial infections. 69 Previous studies have shown that the immune cells carry PRRs on their surface or cytoplasm, allowing them to identify and destroy invading pathogens.70–73 These PRRs activate the immune system in response to recognizing PAMPs, and endogenous chemicals are known as damage-associated molecular patterns (DAMPs).70–72 Different PAMPS found in several types of bacteria, and viruses also can be found in other foreign bodies. 74 Among PRRs, are the membrane-bound TLRs family. 13 TLRs are type I membrane-associated glycoproteins which are members of the superfamily of interleukin-1 receptors (IL-1Rs). 75 TLRs have a modular structure consisting of an extracellular domain comprised of 20–27 leucine-rich repeats (LRR) coupled to an intracellular Toll/Interleukin-1 receptor (TIR) domain.76–78 TLRs are expressed on various innate immune cells, such as dendritic cells, macrophages, natural killer cells, and circulating leukocytes like neutrophils and monocytes, as well as on cells of adaptive immunity like T cells and B cells, and on non-immune cells such as epithelial and endothelial cells and fibroblasts.79, 80 Besides, it is found in different organs like the kidney.9, 13, 81 TLR especially type 4 has been found in the thick ascending limb of the kidney. 82 TLR-4 expression is mostly seen in proximal and distal tubular epithelial cells rather than in intrinsic renal cells such as mesangial, glomerular endothelium, and podocyte.82–85
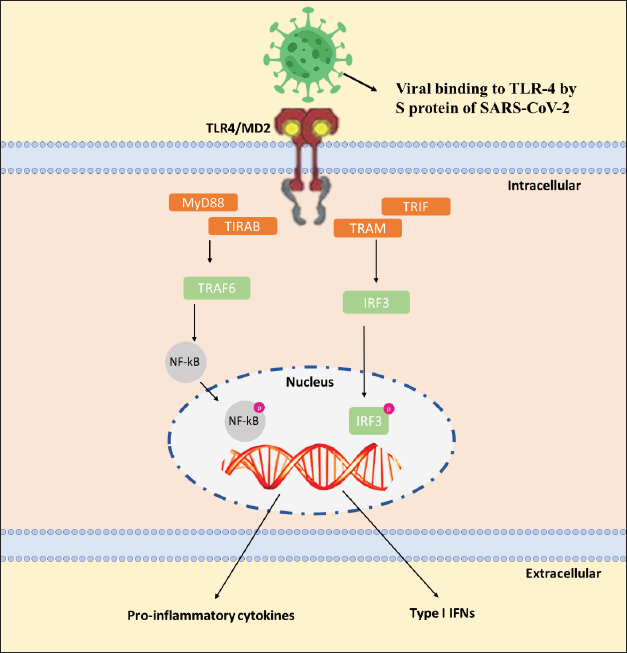
TLR-4 recognizes a wide range of chemicals, including lipopolysaccharide (LPS), the primary molecular component of the cell wall of Gram-negative bacteria. Also, TLR-4 recognize viruses, fungi, and mycoplasma.86–88 Oxidized phospholipids (oxPLs), oxidized low-density lipoprotein (oxLDL), heat shock proteins (HSPs), extracellular matrix (ECM), cathelicidin (LL37), hyaluronic acid, and substance P are examples of DAMPs. These molecules emerge after injury and inflammation and operate as TLR-4 agonists.89, 90 An agonistic ligand interacts with the LRR-containing extracellular domain to begin TLR signaling. For LPS and virus recognition, TLR-4 uses an auxiliary protein called myeloid differentiation protein-2 (MD2). MD2 plays a role in TLR-4 trafficking to the cell surface by binding to TLR-4 within the cell. 91 Cluster of differentiation-14 (CD14), the coreceptor, is also involved in endocytosis and is responsible for transporting LPS to TLR-4.91, 92 The ligand detection by TLRs leads to the structural rearrangement of the extracellular domains. 93 After dimerization, TLR-4 sends signals along two distinct routes: the myeloid differentiation primary response 88 (MYD88)-dependent route and the MyD88-independent route. The phosphorylation of transcription factors like nuclear factor-kappa B (NF-κB) and activator protein 1 (AP-1) occurs due to the activation of interleukin-1 receptor-associated kinases (IRAKs) and TNF receptor associated factor 6 (TRAF6) in MyD88-dependent pathways. After phosphorylation, these transcription factors produce pro-inflammatory cytokine genes by translocating to the nucleus. The MyD88-independent route, also known as the TIR-domain-containing adapter-inducing interferon (TRIF)-dependent pathway, however, includes TRAF3 for the activation of interferon regulatory transcription factor 3 (IRF-3), which promotes the synthesis of type I interferons such IFN α, β. 94
TLRs had an important role in the illness associated with COVID-19, according to research on other COVIDs and COVID-19. According to research by Conti et al., activation of TLRs by the COVID-19 infection may cause an elevation the pro-inflammatory cytokines production such as interleukin-1beta (IL-1β), interleukin-6 (IL-6), and tumor necrosis factor-α (TNF-α) (Figure 2).95–98
In 2020 in silico analysis, Choudhury et al. showed that TLR-4 has the strongest interaction with SARS-CoV-2 spike glycoprotein than any other TLRs. 99 Demonstrates that, of the several TLRs on the surface of cells, the TLR4 receptor is the most likely to be utilized in recognizing molecular patterns from SARS-CoV-2 and triggering inflammatory responses.99, 100 Because their discovery revealed that the human ACE2 entry receptor and the spike glycoprotein form a strong bond, the research seems to have validity. 99 The binding of the spike protein to TLR4 may enhance SARS-CoV-2 entrance into human cells and the initiation of the CS, both of which may have far-reaching effects on several organs. CS is an important complication of COVID-19’s severe phases. 100 TLR-4 is responsible for producing IL-6 and TNF-α, the two main cytokines in cases of severe COVID-19. 97
Shon et al. (2020) assessed the transcriptome profiles of 48 participants, 20 of who served as healthy controls, and 28 of whom were COVID-19 patients (20 of whom had mild/moderate symptoms and 8 had life-threatening ones). The COVID-19 patients showed higher levels of inflammatory signaling molecules in their peripheral blood mononuclear cells (PBMCs), which were mediated by TLR-4. 101 The expression of TLR-4 and downstream signaling markers was significantly upregulated (CD14, MYD88, IRAK1, TRAF6, IRAP, TICAM). 102 Most of the genes involved in the NF-κB signaling pathway (NFKBIA, NFKB1, RELA, and NFKB2) were also considerably elevated, becoming hyperactivated in response to a COVID-19 infection. Based on these findings, the increased inflammatory responses in COVID-19-infected patients are caused by activation of the NF-κB signaling pathway through the TLR-4 receptor.103, 104 These results suggest the molecular etiology of COVID-19 is related to inflammatory signaling generated by TLR-4 rather than viral activity.
Also, TLR-4 is activated by DAMPs generated by lysed or dead cells due to COVID-19, leading to inflammation and fibrosis. According to a review by Andersson et al. from 2020, High mobility group box protein 1 (HMGB1) produced as a DAMP or released by activated immune cells may cause activation of TLR-4 and secretion of pro-inflammatory cytokines. 105 Necrosis and oxidative stress, two circumstances that cause cellular destruction, accelerate the release of HMGB1 from within the cells, which in turn causes the inflammatory response by activating PRRs like TLR-4. 106 TLR-4 may thus contribute to the pathogenesis of COVID-19.9, 107
Cytokine Storm
The proteins released from immune system cells are called cytokines. The cytokines facilitate communication and signaling amongst themselves. Depending on the cytokine and the target cell, a wide range of reactions may be triggered by the cytokine’s receptor binding. Cytokines can exert autocrine, paracrine, or endocrine activity.
Comparable to SARS-CoV-1, SARS-CoV-2 infections have pathogenesis similar to SARS-CoV-1, and the virus’s airway damage strongly correlates with powerful inflammatory responses. 108 Therefore, in addition to viral infection, the host reaction also contributes to illness severity in patients.7, 22, 33 This reaction seems abnormal in certain cases and may affect patients’ immune systems more than the viral damage. 109 The infection sparks a local immunological response with SARS-CoV-2 and lung cell destruction. This reaction attracts macrophages and monocytes, which fight the virus by releasing cytokines and activating adaptive B and T cells immune responses. This process is usually successful in eliminating the infection. However, a defective immune response may emerge, leading to serious lung and even systemic disease in certain instances. 110 One of the key causes of the poor prognosis of severe COVID-19, having multiple organ failures and mortality accompanying serious cases of COVID-19, is CS.6–8
Recently, Huang et al. have confirmed that COVID-19 patients contributed to a rise in pro-inflammatory cytokines and chemokine. 7 Many studies evaluating the cytokine profiles of COVID-19 patients indicated that the CS substantially impacted the onset of ARDS, the adverse prognosis of serious COVID-19, and multi-organ failure. In CSS, the high production of pro-inflammatory cytokine factors like IL-6 can be generated by tissue damage due to the virus.7, 8 Moreover, COVID-19 patients have a massive and significant generation of pro-inflammatory cytokines like interleukins (IL-1β, IL-6, IL-10, and IL-4), interferon γ (IFN γ), TNFα, and chemokines (CXCL10 and CCL2) by immune effector cells precipitate and supports the unusual systemic inflammatory reaction.7, 111–113 As mentioned before, cytokines have a crucial role in inflammation, and for patients with COVID-19, many immune cells are responsible for the production of cytokines. 114 Above all, CS was a major driving factor for a more serious clinical course. According to research, individuals admitted to the ICU with COVID-19 had considerably greater TNF, CCL2, and CXCL10 than those with a less severe infection that necessitated ICU admission. 7 Anti-viral immunity against coronaviruses is initiated by a reaction of T lymphocytes to the virus’ spike protein.
Moreover, T cells can play pro- and anti-roles in developing COVID-19, acting as a double-edged sword. 115 Studies have shown an increase in T helper-2 (Th2)-immune cytokine secretion like IL-4 and IL-10 in SARS-Cov-2 patients compared to SARS-CoV infection. 116 A change in the Th1 to Th2 cytokine balance can lead to chronic infections because the Th2 cytokine suppresses and prevents anti-viral Th1 responses where the inflammation is mainly suppressed as the key effect of IL-4 and IL-10. 117 CS is highly connected with immune response, which will be a reason for ARDS and severe organ failure, which could be responsible for mortality, mainly in serious SARS-CoV-2 infection. 118
During infection, lymphocytes produce inflammatory cytokines to destroy infected cells. 119 Excessive cytokine release may cause AKI in patients having COVID-19 through interacting with kidney-resident cells, which will cause dysfunction of the endothelium, microcirculatory derangement, and tubular damage. 120 The development of COVID-19-induced AKI by CS may result in sepsis, shock, hypoxia, and rhabdomyolysis.23, 121 Due to the disruption of the oxygenated blood supply, increased pressure on the blood vessels in the kidneys’ tubular system, the uncontrollable production of inflammatory mediators and cytokines, and potential renin-angiotensin-aldosterone system activation (RAAS), these effects ultimately cause kidney damage. 122 The imbalance of the RAAS, which is connected to the expression of the ACE2 receptor in the kidneys, causes simultaneous inflammation, fibrosis, and vasoconstriction. 123 Due to increased pro-inflammatory cytokines seen in SARS-CoV-2 patients, which induce immune-directed harm to the glomeruli, it has been proposed that CS causes collapsing glomerulopathy.124, 125
Therapeutic Targeting TLR-4
All of the presented findings agreed with the possibility that SARS-CoV-2 might lead to an increase in the CS and then multi-organ failure. Therefore, controlling numerous mediators might potentially have more positive effects than inhibiting a single mediator. IL-1 receptor antagonists and anti-TNF monoclonal antibodies have been unsuccessful in treating sepsis patients because the other inflammatory signaling pathways remain unregulated.126, 127
Corticosteroids are well-known medications for the treatment of autoimmune and inflammatory conditions, and they are frequently used in the management of COVID-19. 128 They interact with nuclear receptors to inhibit the release of cytokines that cause inflammation. 129 Additionally, steroids lessen HMGB1 release and TLR-4 interaction. 130 A recent meta-analysis of four randomized controlled trial that evaluated prolonged methylprednisolone therapy for ARDS found that the number of ventilator-free days increased (from 7 to 13; p <0.001) while mortality was significantly decreased. 131 Other studies, on the other hand, have been unable to show strong proof of the effectiveness of corticosteroids in reducing ARDS mortality, indicating that glucocorticoid therapy is not required in this situation and may even worsen the clinical course of the illness. For prior coronavirus infections, SARS, and Middle East respiratory syndrome the usage of steroids is still debatable. 132 In a meta-analysis, the researchers determined that the administration of corticosteroids in patients infected with SARS-CoV-2 delayed viral clearance and did not significantly increase survival. Recent evidence also suggests that early, short-course methylprednisolone lowers mortality and progression to respiratory failure, ARDS, and ICU hospitalization. 133 Another Wuhan study on SARS-CoV-2 pneumonia found that patients were given intravenous methylprednisolone had shorter ICU stays and quicker peripheral oxygen recovery. 134 These findings suggest that short-term methylprednisolone therapy should be considered before the onset of ARDS in patients with severe COVID-19 pneumonia. 132 Although there are different sources on corticosteroid use in critically ill patients with COVID-19, the question remains regarding the use of corticosteroids in AKI induced by COVID-19.
The TLR-4 signaling system opens therapeutic targeting and clinical application opportunities because. 135 TLR4 activation is necessary for the body to fight an infection, but too much stimulation might be harmful, particularly later on. Despite this, if the TLR-4 receptor is blocked, other TLRs will still be able to produce some interferons. TLR-4 may be present in endosomes as well as on the surface of the cell. According to the findings of this review, focusing on TLR-4 as a possible therapy for CS in COVID-19 might be beneficial. 100 In the severe phases of COVID-19, administration of a TLR-4 antagonist may protect against the abnormally high TLR4 activation that may otherwise cause a CS and organ failure. Additionally, it can potentially prevent the virus from entering at an earlier stage. It has been found that using TLR-4 antagonists has consistently decreased cytokine and chemokine production, and also ameliorated illness symptoms in small animal models infected with influenza A virus (IAV), Ebola virus (EBOV), respiratory syncytial virus, and dengue virus.136, 137
Based on Choudhury and Mukherjee, it is recommended to target TLRs to fight COVID-19. 99 An exciting range of chemical substances may interact with the TLR4 pathway. Eritoran (E5564) and TAK-242 (Resatorvid) are two of the most well-known TLR antagonists; they attach to the LPS binding pocket on the surface of cells and the TIR domain on the inside of cells, respectively.138–140 TAK-242 inhibits TLR-4’s ability to function by interacting with Cys747 in the intracellular domain, while E5564 binds to the TLR-4/MD2 complex to block TLR-4 activation.138, 141 The molecular weight of TAK-242 is small (360.1), and it has good liposolubility in contrast to E5564, which has poor liposolubility and high molecular weight. Mouse TLR-4 and human TLR-4 are 67% identical and 79% comparable in terms of amino acid composition. Similar inhibition of cytokine production was shown with TAK-242 in primary mice and human monocyte/macrophages.139, 142 Eritoran and TAK-242 have been used to treat inflammation in cancer, rheumatoid arthritis, and other inflammatory illnesses. Since TLR-4 is implicated in COVID-19 pathology, targeting it with antagonists may benefit patients with severe COVID-19 problems. 143 Based on a comprehensive review, TLR-4 antagonists should be used to treat severe COVID-19 infections to minimize inflammation.
Conclusion
A pandemic has been caused by COVID-19, which is highly contagious and has spread quickly over the globe. In patients with COVID-19, kidney impairment, primarily AKI, hematuria, and proteinuria occur. AKI, a severe COVID-19 consequence, greatly impacts the prognosis of patients. Several factors contribute to the pathogenesis of AKI in COVID-19, including the direct cytopathological effects of SARS-CoV-2, renal damage brought on by other multi-organ failures, an unbalanced RAS activation, and pro-inflammatory cytokines induced by the viral infection. Here, we focused on the involvement of aggressive cytokine production and TLR-4 activation as putative underlying mechanisms of SARS-CoV-2-induced kidney damage. There is currently no particular treatment for COVID-19-induced AKI. Therefore, further research is needed to clarify renal involvement and establish the diagnostic, prognostic, and therapeutic strategies used in clinics. Designing new medications to treat the condition requires a thorough understanding of the molecular pathways and key targeted compounds. Blocking viral entry points, including viral receptor(s), viral priming enzymes, and immune response control, are crucial strategies that may be needed to stop the virus and lessen multi-organ dysfunction. Further considerations and studies are needed to help control this pandemic for various medicines that target TLR-4 and prevent the production of cytokines.
Footnotes
Acknowledgments
This review would not have been feasible without the remarkable assistance of my supervisor, Dr. Ahmed Esmat, Department of Pharmacology, Faculty of Medicine, King Abdulaziz University, Jeddah 21589, Saudi Arabia. His energy, intelligence, and meticulous attention to detail have inspired me and kept my work on track.
Authors’ Contributions
M.A.A. managed the literature searches, drafted the manuscript, written the manuscript and designed the figures. M.A. was involved in the planning. All authors discussed and commented on the manuscript. All authors have read and agreed to the published version of the manuscript.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Ethical Approval
Not applicable.
Funding
The author(s) received no financial support for the research, authorship and/or publication of this article.
Informed Consent
Not applicable.