
Abstract
Juvenile dermatomyositis (JDM) is the most common form of idiopathic inflammatory myopathy in children and adolescents. The exact pathogenesis is not clear; however, a combination of several factors, including genetics, immunological abnormalities and environmental factors, has been implicated in the pathogenesis of JDM. The role of Type I interferons in the pathogenesis of JDM has received a lot of attention recently. JDM is characterised by proximal muscle weakness, elevated muscle enzymes, and pathognomonic skin rashes such as heliotrope rash and Gottron’s papules. Main long-term complications include calcinosis, lipodystrophy, and secondary osteoporosis. With the advent of myositis-specific antibodies (MSA), important phenotypic subtypes of the disease are characterised. Treatment of JDM is multidisciplinary. Prolonged immunosuppression with physical and medical rehabilitation is the backbone of therapy. The outcome has improved over the years with early recognition and initiation of immunosuppressive treatment and increasing use of steroid-sparing immunomodulators.
Keywords
Introduction
Idiopathic inflammatory myopathies (IIM) are a diverse group of diseases distinguished by proximal muscle weakness and inflammatory changes in skeletal muscle. IIM has several subtypes, including dermatomyositis, polymyositis, inclusion body myositis, and amyopathic dermatomyositis. 1 Juvenile dermatomyositis (JDM) comprises 85% of all children with IIM.2,3 It typically affects the skin and muscles, but disease manifestations vary and can involve other organ systems. 4
Despite decades of research, the precise cause of JDM remains unknown. Systemic vasculopathy plays an important role in the pathogenesis of JDM. The role of Type I IFNs in the pathogenesis of JDM has received a lot of attention recently. Studies indicate that children with JDM have activation of the Type I IFN pathway (IFNα/β) in their peripheral blood and muscle tissues. 3
Epidemiology
JDM occurs all over the world. Estimated incidence rates of JDM in the United States ranged between 2.5 and 4.1 cases per million children per year. 5 The retrospective surveys conducted in Europe revealed incidences ranging from 1.9 to 2.4 cases per million children per year, respectively. 6 The usual median age of onset is 5.7–6.9 years, and age at diagnosis is 7.4–7.7 years. JDM is more common in girls than boys, with a ratio ranging from 1.6 to 2.5:1. 7
Etiopathogenesis
Genetics, immune abnormalities, and environmental factors influence the pathogenesis of JDM (Figure 1). Family history of autoimmunity in some patients suggests that genetics plays a role. A GWAS study identified SNPs in the MHC class II region of chromosome 6 linked to disease susceptibility.8–13 The HLA region, within the MHC, is highly polymorphic and associated with autoimmune diseases, including JDM. Key haplotypes linked to JDM risk include DRB10301, DQA10501, and HLA-B08, while protective haplotypes include DQA10201, DQA10101, and DQA10102. Additionally, SNPs in CCL21, PLCL1, and BLK, as well as the protective TNF-α 238AG allele, are associated with JDM. Variations in IL-1 genes, such as IL-1α +4845TT and IL-β +3953T, are linked to JDM and may indicate disease severity.14–16
Pathogenesis of Juvenile Dermatomyositis (JDM) Showing Complex Interactions Between Genetic Factors, Immune System Abnormalities, and Environmental Influences. Genetic Risk Factors Include the MHC Class II Region of Chromosome 6, With Specific Haplotypes (e.g., DRB10301, DQA10501, HLA-B08) Linked to Increased Disease Susceptibility. Immune Activation Is Central to JDM Pathogenesis, as Evidenced by Muscle Biopsies That Reveal Lymphocytic Infiltration of B Cells, T Cells, and pDCs (Plasmacytoid Dendritic Cells), Including the Complement System, Which Leads to Endothelial Cell Injury and Elevated IFN Levels.
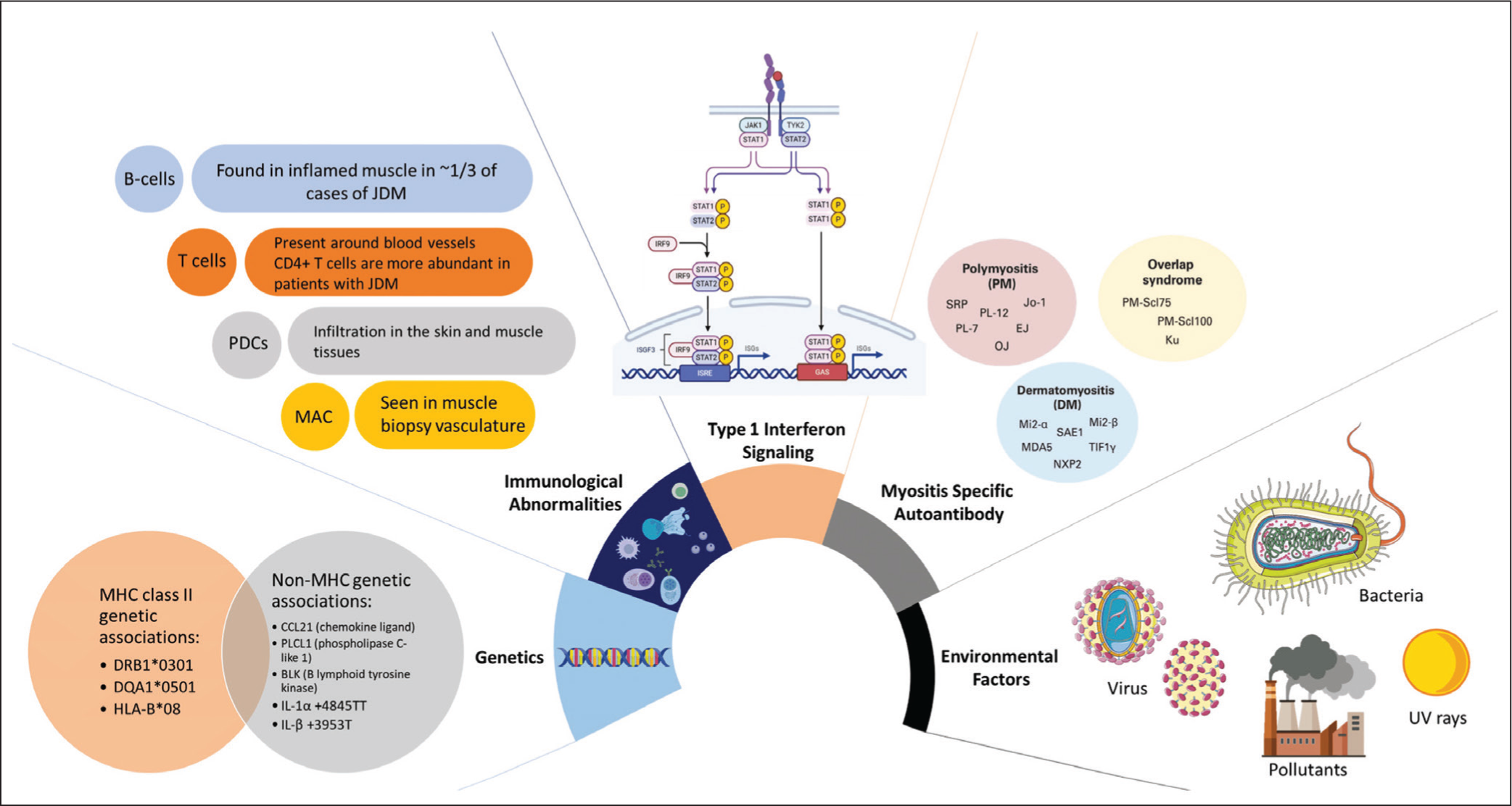
The exact trigger of inflammation in JDM is unclear, but muscle biopsies often show lymphocytic infiltration, MHC class I expression in muscle fibres, blood vessel abnormalities, and muscle damage. Vasculopathy in JDM involves immune activation, including the complement system, leading to endothelial cell injury. Elevated interferon (IFN) levels are linked to vasculopathy and muscle atrophy, with excessive IFN production potentially damaging immune cells and muscle fibres.17–19
In JDM, T lymphocytes (CD4+) and B lymphocytes are found around blood vessels in the muscle, in contrast to polymyositis (PM), where CD8+ T cells dominate. One-third of JDM cases show B cells in the muscle, and over 70% of patients have myositis-specific or associated autoantibodies. Genetic risk alleles are linked to specific autoantibody patterns, such as DQB102 and HLA-DRB103 with anti-PM-Scl, or DPB10101 and DRB10301 with anti-Jo-1 autoantibody.20–24
Rituximab, a B-cell-depleting treatment, showed faster recovery in JDM patients compared to adult DM patients, suggesting B cells play a critical role in the disease pathogenesis. T lymphocyte types in muscle tissue vary by condition: CD8+ T cells dominate in PM and IBM, while CD4+ T cells are more common in DM.25–27
In addition to immune-mediated mechanisms, viral infections such as coxsackievirus, parvovirus B19, and SARS-CoV-2 have been reported as potential environmental triggers of JDM. Furthermore, non-immune mechanisms, including mitochondrial dysfunction and endothelial injury, contribute to chronic vasculopathy and calcinosis.13–17
Calcinosis
Calcinosis, seen in 20%-40% of JDM patients, involves abnormal calcium salt deposition, most commonly as dystrophic calcification. It is associated with prolonged inflammation, elevated muscle enzymes, and severe manifestations like dysphagia, weakness, and joint immobility.28–31 Morphologically, calcinosis may be superficial, nodular, or tumoral (Table 1).30–43 Calcinosis is primarily a form of dystrophic calcification that occurs in the setting of chronic tissue inflammation and necrosis, despite normal serum calcium and phosphate levels. Persistent inflammatory activity, mitochondrial dysfunction, endothelial injury, and disordered collagen fibrils act as nucleation sites for calcium-phosphate deposition. Cytokines such as TNF-α and IL-1, together with macrophage activation, perpetuate local inflammation and mineralisation. Genetic predispositions (e.g., TNF-α 308A allele, anti-NXP2 antibody positivity) further increase risk. In addition, mitochondrial calcium efflux and endothelial matrix protein deposition have been implicated, linking calcinosis with chronic vasculopathy in JDM.34–43

Clinical Features
The clinical presentation of JDM varies depending on the myositis-specific antibodies (MSA) subtype. Almost all JDM patients exhibit some degree of muscle weakness. JDM can present with an acute or insidious onset; the median time from symptom onset to diagnosis is 3–7 months. 3
Constitutional Symptoms
Fever, weight loss, anorexia, and fatigue may be seen in JDM patients and vary according to the severity of the presentation. Some patients exhibit arthritis and nonspecific lymphadenopathy. 4
Muscle Involvement
Muscle involvement in JDM is typically characterised by symmetrical proximal muscle weakness, present in 95% of patients at the time of diagnosis. 3 Patients report difficulty climbing upstairs, getting up and down, dressing, combing hair, lifting the head when lying flat on the bed, choking while drinking liquids, and a voice change. Muscle involvement can be measured with a variety of scores.
Muscle groups commonly involved in JDM are neck flexors, shoulder abductors, hip flexors, and hip extensors. Polymyositis, on the other hand, primarily affects the hip flexors, hip abductors, knee extensors, and ankle plantar flexors, while dermatomyositis patients primarily exhibit weakness in wrist flexors, hip flexors, and hip extensors. 44
Validated muscle testing tools, such as the Manual Muscle Test (MMT) and the Childhood Myositis Assessment Scale (CMAS), should be used to formally examine muscle strength. Both tools have been proven to be accurate and helpful in evaluating muscular strength during the diagnostic and follow-up phases. 45
In severe cases (CMAS <15), patients experience widespread weakness affecting both proximal and distal muscles. The palatal and pharyngeal muscles become involved, resulting in dysphagia, dysphonia, aspiration, and food reflux. Respiratory muscle weakness may develop, leading to respiratory failure. Patients who present with muscle weakness, difficulty swallowing, nasal intonation, tachypnoea, or shortness of breath should be closely monitored for respiratory failure and aspiration, which may be life-threatening. Later in the disease, muscle atrophy may develop. 3
Dermatological Involvement
The characteristic rash of JDM includes Gottron’s papules and heliotrope rash (violaceous periorbital oedema) (Figure 2). Gottron’s papules are one of the pathognomonic symptoms of JDM. These are small, violaceous, flat-topped papules that appear on knuckles, dorsal wrists, knees, and elbows. 46 Inverse Gottrón’s papules have also been reported, particularly in the adult Asian population. 47 These are flat-topped papules that range in size from linear to triangular. They can be found on the palmar surface of the mid-digits and the skin that covers the joints. Other lesions seen in JDM include shawl sign, rash, including the upper chest region, facial rash, photosensitive erythema, truncal erythema, oral ulcers, Holster sign (rash over the lateral aspect of the thighs), ragged cuticles with nail fold changes, calcinosis cutis and more. Malar rash in JDM, unlike SLE, tends to involve nasolabial folds also which is one of the soft clinical pointers to differentiate lupus from JDM. These abnormalities are present in around 75% of patients presenting with JDM and may precede or follow the onset of muscle involvement by months to years. 44
Representative Images of the Different Cutaneous Manifestations in Juvenile Dermatomyositis in Different Patients in Our Cohort. (A) Heliotrope Rash; (B) Gottron’s Papules; (C) Inverse Gottron’s Papules; (D) Calcinosis; (E) Nailfold Capillary Changes; (F) Lipodystrophy.
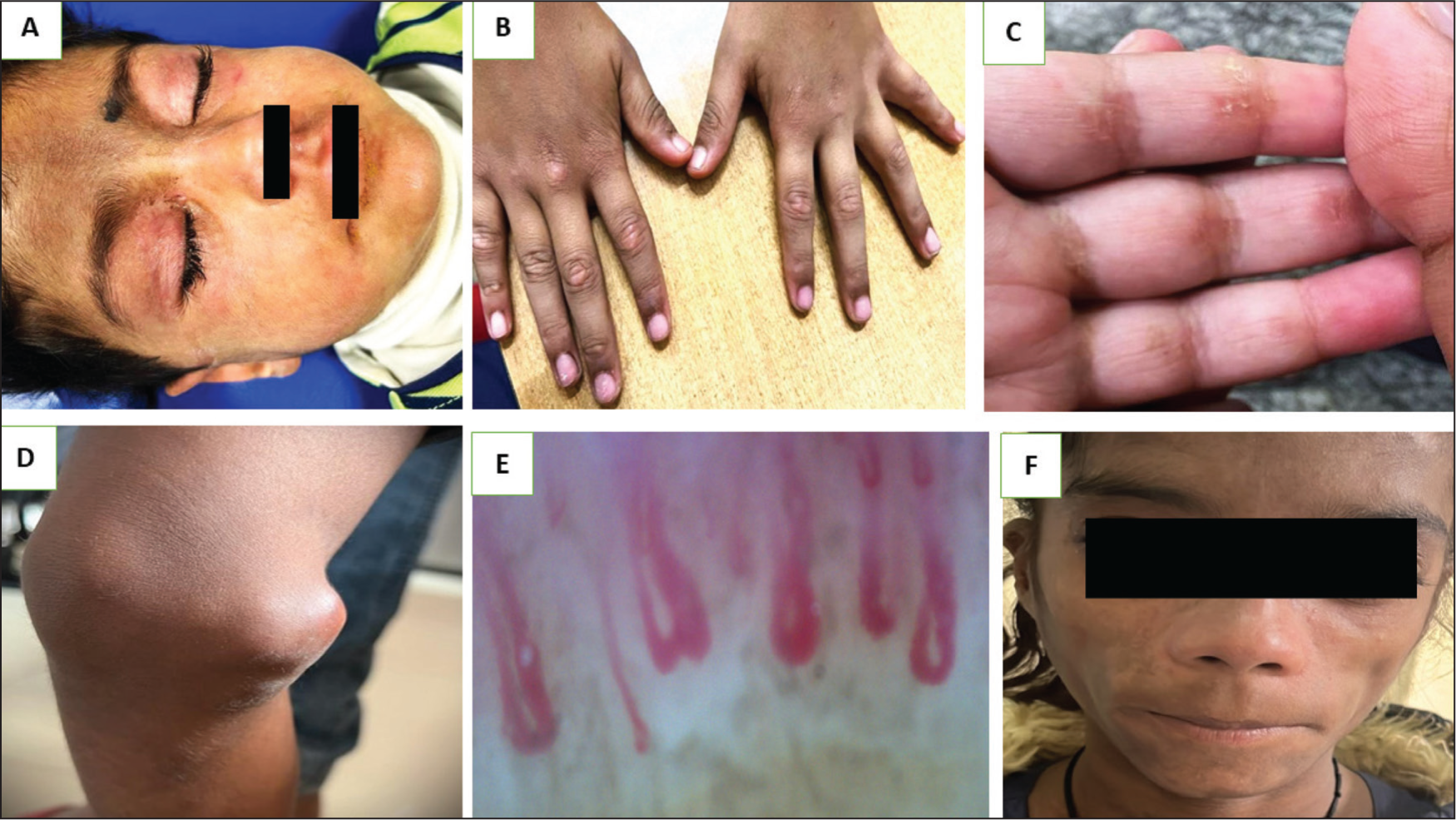
Patients with JDM may also experience facial and periorbital oedema. Subcutaneous oedema may occasionally be the only sign of the illness and can be misdiagnosed as nephrotic syndrome.48,49
Skin manifestations are caused by underlying micro-vasculopathy, and 20% of patients develop skin ulcers, indicating a severe course of disease. Skin ulcers are caused by vasculopathy, characterized by hypoxia and small vessel ischaemia. Skin ulceration may also indicate vasculopathy in other internal organs, which can lead to ulcerative changes in the intestine, resulting in haemorrhage, pneumatosis intestinalis, or even perforation. 50
Nail fold capillaroscopy (NFC), which allows for the visualisation of small periungual capillary changes, can detect cutaneous micro-vasculopathy early. Changes observed include dilation, haemorrhages, occlusion, capillary dropouts, and increased tortuosity. NFC changes are present in 68%-91% of patients at the time of diagnosis. Nailfold capillary density is a widely used non-invasive tool to indicate disease activity and for follow-up of disease.51,52
Calcinosis usually happens at pressure points like the knees, buttocks, elbows, and digits. Calcinosis can be present at the time of presentation or years later, but it usually appears within a few years of disease onset. Calcinosis can lead to long-term disability by causing skin ulcers, neuropathy, and joint contractures. 53
Lipodystrophy affects 8%-14% of children with JDM. (Figure 2) It causes a progressive decrease in subcutaneous and visceral fat in general. It has been observed to cause metabolic syndrome-like symptoms such as dyslipidaemia, insulin resistance with acanthosis nigricans, and diabetes. 54
Pulmonary Involvement
Pulmonary involvement in JDM varies according to ethnicity and MSA subtypes. Respiratory system involvement may manifest as dysphonia, dyspnoea at rest, abnormal pulmonary function tests, interstitial lung disease (ILD), and pneumothorax. Lung involvement indicates a poorer prognosis, with adult DM being five times more fatal than JDM. CADM (clinically amyopathic dermatomyositis) and high levels of anti-MDA5 and anti-synthetase antibodies are risk factors for rapidly progressing ILD. HRCT lung (abnormal in 37% of patients) and pulmonary function tests (reduced in more than 50% of patients) reveal either a restrictive or obstructive pattern.55,56
Cardiovascular Involvement
Cardiac involvement in JDM is frequently underestimated, and it may be an indicator of poor prognosis. It varies between 9% and 72% in different cohorts. Cardiac dysfunction in JDM can occur both acutely and chronically. The most common acute complications are congestive heart failure (CHF), myocarditis, arrhythmia, and complete heart block. However, because there are no obvious clinical manifestations, these go unnoticed. Rare life-threatening complications like pulmonary arterial hypertension can also be seen. Positive anti-SRP and anti-Jo 1 autoantibodies have been linked to an increased risk of cardiovascular abnormalities. Long-term follow-up studies in JDM have revealed an increased incidence of hypertension, atherosclerosis, coronary artery disease, and metabolic syndrome in adolescence and adulthood. Monitoring of body mass index, blood pressure, and laboratory testing of fasting glucose and lipid profile may aid in the identification of metabolic syndrome in children with JDM. Steroid-free medications, regular exercise, and a healthy diet may all help to reduce long-term cardiac morbidity. The current use of multimodality imaging, such as stress echocardiography, contrast-enhanced echocardiography, cardiac magnetic resonance imaging, and positron emission tomography, has improved the diagnostic yield of subclinical heart disease during the acute and chronic stages of JDM.56,57
Gastrointestinal Manifestations
GI involvement affects between 18% and 44% of JDM patients. Dysphagia, gastroesophageal reflux, delayed gastric emptying, bowel dysmotility, stomach vessel inflammation, ulcers, bleeding, and perforation are the GI manifestations in JDM. The underlying pathology is thought to be occlusive thrombosis and vasculitis. Persistent abdominal pain in a JDM child may indicate the presence of an underlying GI vasculopathy, such as ischaemic ulcers and perforation.58,59
Laboratory Features
Elevation of at least one muscle enzyme, like creatine kinase (CK), lactate dehydrogenase (LDH), aspartate aminotransferase (AST), alanine aminotransferase (ALT), or aldolase, is seen in 80%-96% of JDM patients.60,61 CK level may be elevated in only 50% of patients with JDM at the time of diagnosis. 62 Despite active disease, several patients present with normal muscle enzyme levels due to late presentation and muscle wasting. Presence of elevated muscle enzymes at disease onset does not predict disease outcome. A complete blood count is usually normal, but in some cases, leucocytosis and anaemia are present. Erythrocyte sedimentation rate (ESR) and C-reactive protein (CRP) levels may be elevated during active disease, but active disease can also be seen with normal ESR and CRP. Galectin-9 and CXCL10 are biomarkers that have been tested in multiple cohorts and proposed to differentiate between JDM patients with active disease and those in remission. Even in the absence of elevated CK levels, persistently high or rising levels of these biomarkers indicate an impending flare, which can occur several months before clinical symptoms appear. 45
Autoantibodies in JDM
Myositis antibodies consist of both myositis-specific autoantibodies (MSA) and myositis-associated autoantibodies (MAA). MSAs are found only in JDM patients, whereas MAAs are found in patients with other autoimmune conditions linked to myositis. MSAs are found in approximately 40%-60% of children with JDM. The presence of MSA suggests myositis, and because of their associations with specific clinical features, these antibodies may predict clinical phenotypes and outcomes in JDM.63,64
MSA
Anti-p155/140 or anti-TIF1 autoantibodies are antibodies that target the 155-kDa protein TIF-1 gamma (transcriptional intermediary factor 1 gamma) and the 140-kDa protein TIF-1 alpha, respectively. They are detected in 17.9%-35% of JDM patients, making them the most common autoantibody. Anti-TIF-1gamma autoantibody was linked to more severe cutaneous manifestations such as extensive photosensitive erythema, Gottron’s papule, shawl sign, and the development of lipodystrophy, skin ulcers, and oedema. It is associated with higher UV indices and polycyclic disease progression. Anti-TIF-1 gamma antibodies have been linked to cancer in adults with dermatomyositis. However, the same observation has not been noted in children with JDM.63–65
Anti-MJ/NXP2 autoantibody is most seen in JDM (18%) and is a poor prognostic factor. These patients have a younger age at onset and a lower chance of remission at two years. This autoantibody has been linked to an increased risk of calcinosis, dysphonia, atrophy, joint contractures, severe muscular weakness, gastrointestinal ulcers, and bleeding. 64
Anti-MDA-5 autoantibody is directed against the melanoma differentiation-associated gene 5, also known as CADM-140. This antibody is more common in the Asian population. While the Japanese cohort of JDM had a prevalence of anti-MDA-5 of 23.8%-33%, the UK cohort of JDM had a prevalence of 7%. Clinically, these patients had skin and oral ulcers, amyopathic forms, and arthritis. The amyopathic form is less common in JDM, with muscle weakness being more frequently observed in JDM (93%) compared to DM (32.8%). 66 This autoantibody is linked to a higher risk of ILD, which is rapidly progressive.63,65,67
Anti-Mi-2 autoantibodies against the nucleosome remodelling and deacetylase complex are a specific marker for JDM, though less commonly seen (4%-10%). It is strongly associated with dermatological manifestations in adult DM but not in JDM. This antibody has a better prognosis, with milder muscle involvement and a lower risk of ILD and cancer. These patients had a lower mortality because they were more responsive to standard treatment.63,64
Anti-aminoacyl-tRNA synthetase (Anti-ARS) autoantibodies are antibodies against aminoacyl-tRNA synthetases such as JO-1, OJ, EJ, KS, PL-7, Zo, PL-12, and Ha. These autoantibodies are more common in adult DM than in JDM.
Anti-SAE antibodies are one of the rarest MSAs reported in children with JDM. They are usually reported in adults and have been shown to have an association with malignancy.64,68
MAA
They are not so specific for myositis. They are seen in patients with JDMS overlapping with other connective tissue disorders, mainly scleroderma. Most common MAAs include anti-Ro (SSA), anti-La (SSB), anti-PM-Scl, anti-U1-RNP, and anti-Ku autoantibodies. Anti-SSA (Ro60 and Ro52) is seen in 6% of JDM and 14% of juvenile myositis. Anti-Ro52 autoantibody is seen associated with anti-Jo-1 and anti-MDA5 autoantibodies. A combination of anti-Ro52 and anti-Jo-1 is shown to be associated with ILD and poorer prognosis. 64
Radiological Investigations
Ultrasound of Muscles
Muscle ultrasonography is a useful, low-cost, non-invasive modality. Oedema, muscle atrophy, and/or fatty infiltration can cause increased muscle echogenicity and decreased muscle volume, as seen on ultrasound. Calcification can be seen clearly on ultrasound as echogenic foci with posterior acoustic shadowing. The ultrasound findings appear to be highly correlated with disease activity; patients with no symptoms or signs of active disease typically have a normal muscle sonographic appearance, whereas patients with active disease have different patterns of muscle changes. 69
MRI of Muscle
MRI has helped diagnose JDM and monitor myositis activity. This imaging modality does not expose children to ionising radiation. Muscle disease can be evaluated without the use of intravenous contrast material. MRI with T2-weighted imaging and fat suppression sequences reveals soft tissue oedema and active disease. Short tau inversion recovery (STIR) sequences help to visualise inflammatory change because increased signal intensity in affected tissues indicates the presence of oedema. T1-weighted sequences can detect muscle atrophy and fatty infiltration in chronic disease. Because proximal muscles are commonly affected, imaging of the pelvic and thigh musculature is usually performed. MRI of the thighs also aids in identifying sites of active disease for muscle biopsy, reducing false-negative results. 45 Some centres prefer whole-body MRI to determine the distribution and magnitude of inflammatory changes throughout the body. Even if muscle inflammation is clinically undetectable during physical examination, patients may exhibit signs of muscle inflammation in their distal limbs. STIR whole-body sequences can also detect subcutaneous tissue and myofascial abnormalities that are otherwise undetectable. Changes in subcutaneous fat signal intensity in the pelvis and thigh are associated with a more aggressive chronic disease course and may predict future calcinosis. When clinical assessment and laboratory parameters are ambiguous, MRI can effectively determine whether a patient is experiencing a disease flare. 70
Treatment
JDM is difficult to treat due to its rarity, heterogeneity of presentation, and a lack of adequate controlled trials. JDM requires a multidisciplinary approach that includes paediatric rheumatologists, physiotherapists, occupational therapists, specialist nurses, dieticians, social workers, and other specialists. The treatment objectives are to reduce inflammation, avoid treatment-related complications and long-term disease sequelae, and improve quality of life.45,71 Evidence is mainly from small case-controlled studies, except for two randomised trials. In 2010, the Childhood Arthritis and Rheumatology Research Alliance (CARRA) recommended a treatment plan for moderately severe JDM for the first two months, including intravenous methylprednisolone followed by oral prednisolone or high-dose oral prednisolone with MTX ± IVIG. The only randomised trial, conducted by PRINTO (2006–2011), found that combining steroids with MTX was most effective and safe. The primary treatment consists of high-dose corticosteroids and steroid-sparing agents (commonly MTX). 50 Primary intensification of therapy with cyclophosphamide is indicated for patients with severe disease: skin ulceration, severe muscle disease with dysphonia/dysphagia, gastrointestinal involvement, or ILD 45 (Figure 3).
Algorithm Showing the Therapeutic Regimen for Juvenile Dermatomyositis.
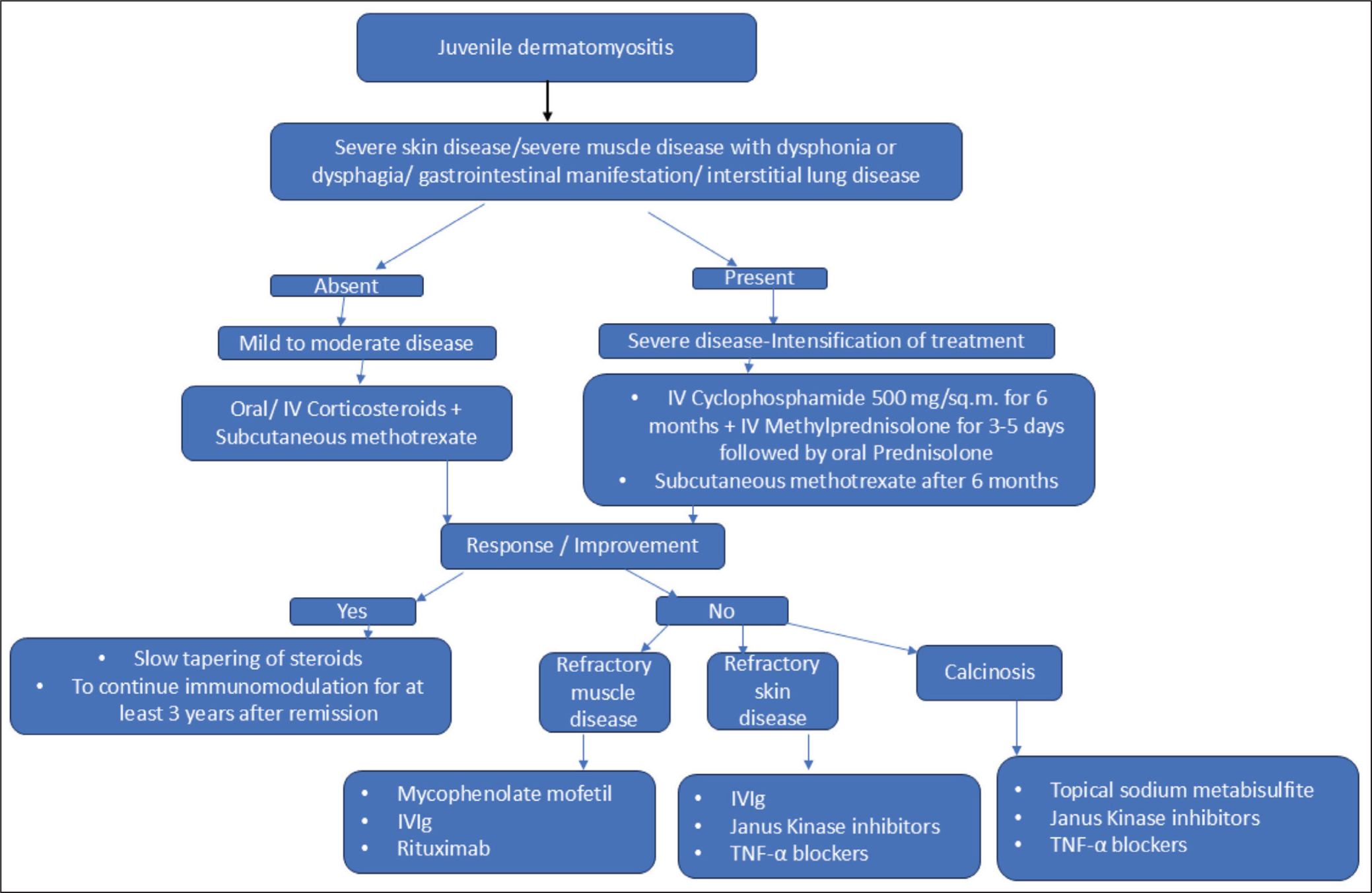
Corticosteroids
They are the central and first-line treatment for JDM patients. Because of steroid treatment, mortality has decreased from 30% to 2%-3%. The initial route of therapy, oral versus intravenous, is still debated, but some argue that IVMP (intravenous methylprednisolone) is preferable for active JDM patients because intestinal vasculopathy reduces oral prednisolone absorption due to decreased enteral blood perfusion. The IVMP dose is 15–30 mg/kg/day, with a maximum dose of 1g for moderate and severe cases. The length of steroid therapy is typically between 4 and 24 months, but can vary depending on the patient’s response to the treatment.45,46 Regarding the length of treatment, recently, the (PRINTO) Paediatric Rheumatology International Trial Organisation evidence-based proposal for steroid tapering or discontinuation recommended reducing the steroid dose gradually to 0.2 mg/kg day over 6 months to lessen long-term steroid exposure and its side effects. 72
Conventional Disease-modifying Anti-rheumatic Drug (DMARDs)
MTX is one of the most commonly used drugs, and it has now become the first-line treatment for JDM. The addition of MTX reduced cumulative steroid exposure while also resulting in higher growth velocities, increased BMI, and fewer cataracts. Combining steroids with MTX resulted in better remission rates than monotherapy alone. The use of CSA in JDM is limited due to the higher side effects, but it is still an option in JDM for patients who have persistent MTX side effects. 72 Hydroxychloroquine is most used to treat cutaneous manifestations of JDM. Cyclophosphamide is thought to be beneficial in refractory cases with significant organ involvement, such as gastrointestinal, lung, skin ulcers, and muscle disease. Mycophenolate mofetil has been used to treat resistant and refractory cases, with promising results including increased muscle strength and the ability to wean off steroids. 73
Intravenous Immunoglobulin
IVIG is regarded as a second-line treatment option, particularly in severe or refractory cases after steroids and MTX. Most reports recommend monthly IVIG administration at a dose of 1–2 g/kg, with a maximum of 70 g from the onset of disease, while others recommend administering once every two weeks for the first three to five times before beginning monthly administration.74,75
Biological Agents
RTX (rituximab) is a monoclonal antibody that targets CD20+ B cells. A trial in adult and juvenile refractory DM patients treated with RTX demonstrated a good steroid-sparing effect, with 83% meeting the criteria for improvement. JDM (as opposed to adult DM) and anti-ARS were found to respond more effectively when treated with RTX. A randomised controlled trial found that B-cell depleting therapy can help patients with anti-Jo-1 and anti-Mi-2 positivity. 76
Anti-TNF agents, such as infliximab and adalimumab, have been shown to improve muscle strength and calcification. A large study of JDM found significant improvement in muscle and skin involvement with anti-TNF drugs. 77
Abatacept has been shown to improve calcinosis and stop its progression, control skin and muscle inflammation, and reduce corticosteroid use. 78
In refractory cases, JAK (Janus kinase) inhibitors and anifrolumab (Type 1 interferon inhibitor) are a relatively new treatment option. IFN (interferon) Type I play an important role in disease pathogenesis, and JAK helps to upregulate IFN-stimulated genes by acting downstream of IFN.79,80 JAK-STAT pathway inhibitors, such as tofacitinib and ruxolitinib, are shown to be beneficial in children with JDM. 79 In a case series published in 2020, 25 JDM children with refractory disease were treated with JAK inhibitors, with 66.7% (16/24) having complete rash resolution and 96% (24/25) showing improvement with therapy. 79
Ultraviolet (UV) exposure frequently aggravates the skin manifestations. For all JDM patients, it is advised to avoid sun exposure and to regularly apply sunscreen with an SPF of 30 or higher that protects against both UVA and UVB rays.
In addition to drug therapy, non-pharmacological care forms a critical component of management. Nutritional support (adequate protein and vitamin D/calcium supplementation), structured physiotherapy protocols (including stretching and strengthening exercises to prevent contractures and maintain mobility), and psychosocial support for children and families are integral to improving quality of life and long-term outcomes. 45
Management of Calcinosis
Calcinosis remains one of the most challenging complications of JDM. Early and aggressive suppression of inflammation with corticosteroids and immunosuppressants is the most effective preventive strategy.81–85 Conventional metabolic agents such as sodium thiosulfate, bisphosphonates, and probenecid can promote regression of deposits in some patients, but responses are often partial and slow. Biologic therapies have expanded the treatment options: TNF-α inhibitors have shown benefit in more than half of refractory cases, while rituximab has produced variable results with generally modest improvement. JAK inhibitors are particularly promising, with rapid and sometimes complete resolution reported in small case series.86–89 Abatacept and tacrolimus have also been effective in selected refractory cases, particularly for cutaneous and ulcerative disease. Surgical excision may be useful for localised, symptomatic calcinosis causing severe pain, recurrent ulceration, or nerve compression, though recurrence is a concern. Even with these strategies, calcinosis often persists, highlighting the unmet need for targeted therapies. Long-term follow-up and individualised treatment planning are essential, as no single approach is universally effective. Ultimately, prevention through early recognition and tight disease control remains the best strategy to minimise the burden of calcinosis in JDM.90–94
Non-pharmacological treatments include mainly surgical excision, which can be effective for recalcitrant calcinosis that involves disability due to severe pain, recurrent ulceration, and nerve compression. Surgical treatment of symptomatic lesions in patients with calcinosis had favourable results, with an experience of partial improvement and complete resolution after surgical removal. However, not all lesions are accessible to surgery in these patients.
Challenges in Resource-limited Settings
In many low- and middle-income countries, children with JDM face delays in diagnosis, limited access to advanced diagnostics, and high costs of biologics or IVIG. In these settings, low-cost alternatives such as methotrexate, hydroxychloroquine, and azathioprine remain the backbone of therapy (Table 2). Their use in real-world Indian practice continues to provide meaningful disease control despite restricted access to newer biologics.
Outline of Management of Patients with JDM.

Outcome
The disease course varies. One-third of patients have a single episode of disease and then achieve remission without relapse (so-called monocyclic course). Between 3% and 30% of patients experience a polycyclic course with more than one remission and relapse; the disparity in estimates for how frequently this occurs is most likely due to different definitions of remission. Despite treatment, approximately 30%-60% of patients still have active disease without remission (chronic course). The median time to remission is 4.7 years. JDM patients with a higher disease activity score at diagnosis are more likely to develop a chronic condition. Musculoskeletal involvement improves more rapidly than skin involvement. The persistence of rash (e.g., Gottron papules) 3 months after diagnosis, as well as abnormal NFC, predicts a longer time to remission.95–97
The long-term outcome of JDM patients has significantly improved over time, mostly due to the introduction of corticosteroid treatment and the early initiation of adjunctive immunosuppressive therapy in the last few decades. The most recent studies show that the mortality rate has dropped from more than 30% to less than 5%. There has also been a noticeable improvement in functional outcomes. However, some patients continue to experience disease activity or damage. 45
In JDM, death can be caused by gastrointestinal vasculitis, which can result in massive gastrointestinal bleeding or intestinal perforation, progressive pulmonary disease, progressive unresponsive myositis, cardiovascular events, or superimposed infections. Delays in diagnosis and treatment, late referrals, poor drug adherence, severe muscle weakness at presentation, and infection are all risk factors for mortality. 98
To conclude, JDM is the commonest form of IIM in children. Vasculopathy and an elevated type-1 interferon signature play an important role in disease pathogenesis. Early diagnosis, prompt recognition of complications, and timely institution of appropriate immunomodulatory therapy are important for optimal outcomes.
Footnotes
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship and/or publication of this article.
Ethical Statement
This is a review article, and therefore, does not need approval from the Institute Ethics Committee.
Funding
The authors received no financial support for the research, authorship and/or publication of this article.
Patient Consent
Informed consent was obtained from the parents of the children whose clinical images have been used in this manuscript.