
Abstract
Background
Sepsis-induced pulmonary injury poses a significant challenge in critical care due to its high morbidity and mortality rates.
Objectives
This study explores the potential of uridine to mitigate sepsis-induced pulmonary damage, specifically targeting the activation of the Kelch-like ECH-associated protein 1 (Keap1)-nuclear factor erythroid 2-related factor 2 (Nrf2) signaling pathway.
Materials and Methods
Both in vitro and in vivo approaches were utilized, incorporating MH-S cell lines exposed to lipopolysaccharide (LPS) to simulate inflammatory conditions and cecal ligation and puncture (CLP)-induced sepsis model in mice. The effects of uridine were evaluated on survival rates, pulmonary damage, bacterial colonization, as well as the expression of NRF2, KEAP1, heme oxygenase-1 (HO-1), and inflammatory cytokines.
Results
Uridine treatment led to elevated levels of Nrf2 and HO-1, reduced KEAP1 expression, improved survival rates, decreased pulmonary damage and bacterial presence, and modulation of the Keap1-Nrf2 pathway. adeno-associated virus-mediated Nrf2 overexpression alleviated lung injury. The NRF2 inhibitor ML385 counteracted the beneficial effects of uridine, underscoring the critical role of the Keap1-Nrf2 pathway in these outcomes.
Conclusion
Activation of the Keap1-Nrf2 pathway by uridine holds promise as a therapeutic strategy to enhance outcomes in sepsis-induced pulmonary damage. Further investigation is warranted to assess the therapeutic potential of uridine in sepsis management and to elucidate the underlying mechanisms of its protective effects.
Introduction
Sepsis is a serious and frequently fatal illness that arises from an overreaction of the immune system to an infection (Cecconi et al., 2018; Lelubre & Vincent, 2018). It causes widespread inflammation, organ failure in several systems, and high death rates worldwide (Cecconi et al., 2018). Particularly vulnerable to sepsis-related harm is the respiratory system (Park et al., 2019), which can often result in acute lung injury (ALI) and acute respiratory distress syndrome (ARDS), which are the main causes of morbidity and death in critically sick patients (Xu et al., 2015). Supportive treatments, including careful fluid control and mechanical ventilation, are the mainstay of current clinical therapy for ALI/ARDS (Gorman et al., 2022). However, the prognosis for many people with ALI/ARDS is dire due to the paucity of effective therapy alternatives (Liu et al., 2022; Wick et al., 2021). The need for novel therapeutic approaches is underscored by the pressing need to find interventions that address the underlying pathophysiological processes of sepsis-induced pulmonary impairment, even in the face of advances in critical care and supportive therapies.
Uridine is a fundamental pyrimidine nucleoside that is made up of uracil and ribose. It is essential for many biological activities, such as the creation of glycogen and genetic materials (Connolly & Duley, 1999; Zhang et al., 2020). Because of its cytoprotective and anti-inflammatory qualities, uridine—the main nucleoside in human blood—has become a viable therapeutic target (Chenna Narendra et al., 2015). Uridine has been shown in recent research to modify cellular responses to stress and damage, indicating that it may be useful in reducing oxidative stress and inflammation (Adant et al., 2022; Jeengar et al., 2017; Le et al., 2013). These characteristics make uridine a promising drug in treating the intricate pathophysiology of sepsis-induced lung damage, where oxidative damage and unchecked inflammation are major contributors (Mironova et al., 2018).
A crucial transcription factor with a leucine zipper motif is nuclear factor erythroid 2-related factor 2 (Nrf2) (Zgorzynska et al., 2021). To encode the enzymes essential for detoxification and antioxidant defense, Nrf2 activation increases the transcription of genes regulated by the antioxidant response element (Wu et al., 2023). Kelch-like ECH-associated protein 1 (Keap1) normally inhibits Nrf2, which makes it easier for it to be ubiquitinated and then degraded by proteases (Wang et al., 2023). At the forefront of cellular defenses against oxidative stress and inflammation is the Keap1-Nrf2 signaling pathway (Yamamoto et al., 2018). Changes in Keap1 brought on by oxidative stress result in nuclear translocation and Nrf2 stabilization, which in turn trigger the transcription of protective genes (Baird & Yamamoto, 2020). Therefore, focusing on this route offers a potential treatment option for illnesses marked by inflammation and oxidative stress, such as lung damage brought on by sepsis.
This work aims to clarify if uridine is a therapeutically effective means of reducing lung damage caused by sepsis by activating the Keap1-Nrf2 signaling pathway. By elucidating how uridine impacts this protective axis, we hope to further the creation of specific treatments for lung harm brought on by sepsis. Our new study looks at uridine as a modulator of the Keap1-Nrf2 pathway, revealing a new treatment strategy. Our findings fill a significant gap in the search for viable therapeutic approaches, given the present paucity of effective therapies for sepsis-induced lung damage and uridine’s capacity to control inflammatory and oxidative responses.
Materials and Methods
Reagents and Chemicals
MCE (China) provided the uridine. Sigma-Aldrich (St. Louis, MO, USA) provided lipopolysaccharide (LPS) to create inflammatory conditions. To assess cell viability, Elabscience (Wuhan, China) provided the Cell Counting Kit-8 (CCK-8) assay kit. Additionally, ML385—a particular NRF2 inhibitor—was acquired from MCE (China). Penicillin, streptomycin, fetal bovine serum (FBS), and RPMI1640 culture media were among the essential cell culture consumables provided by Invitrogen (Carlsbad, CA, USA) and Gibco (Grand Island, New Zealand). R&D Systems (USA) provided mouse enzyme-linked immunosorbent assay (ELISA) kits, which are essential for measuring levels of interleukin (IL)-6, IL-1β, and tumor necrosis factor-alpha (TNF-α). Lastly, secondary antibodies conjugated with horseradish peroxidase (HRP) and antibodies targeting KEAP1, NRF2, heme oxygenase-1 (HO-1), and β-actin were acquired from Abclonal (China).
Cell Culture and Treatment
Murine alveolar macrophage MH-S cells were carefully cultivated at 37°C in a 5% CO2 humidified environment using RPMI-1640 media supplemented with 10% FBS, 1% penicillin, and streptomycin. Cells were seeded onto six-well plates at a density of 5 × 105 cells/well for the experiment, and they were left to adhere for 24 hours. After that, cells were exposed to 200 ng/mL of LPS for 6 hours to mimic bacterial infection. To test uridine’s protective effects, 150 µM of the compound was given 2 hours before LPS exposure. The vehicle (phosphate-buffered saline (PBS)) was administered in an equivalent volume to the control groups.
CCK-8 Assay
Following the manufacturer’s instructions, the CCK-8 assay assessed cell viability. Posttreatment, cells were incubated with the CCK-8 solution for 1 hour at 37°C. A microplate reader measured the absorbance at 450 nm, enabling the quantification of cell viability compared to the untreated control group.
Animals
The study involved adult C57BL/6 male mice, aged 8–10 weeks, and weighing between 20 and 25 g, housed under specific pathogen-free (SPF) conditions with a 12-hour light/dark cycle and provided ad libitum access to food and water. The Institutional Animal Care and Use Committee approved all experimental procedures, adhering to the Guide for the Care and Use of Laboratory Animals.
Animal Experiments
Sepsis induction in mice was achieved through the cecal ligation and puncture (CLP) procedure (Rittirsch et al., 2009). Mice were anesthetized with isoflurane, followed by a midline laparotomy to expose the cecum, which was then ligated and punctured twice with a 22-gauge needle, replaced into the abdomen, and the incision was closed. Postoperatively, mice were resuscitated with 1 mL of sterile saline subcutaneously. Uridine at a dose of 15 mg/kg, or saline as a control, was administered intraperitoneally immediately after the surgery. The animals were euthanized 24 hours post-CLP, and tissues were harvested for subsequent analysis.
Histopathological Analysis of Lung Tissues
Lung tissues were fixed in 4% formalin, embedded in paraffin, and sectioned at 5 µm. Sections were stained with hematoxylin and eosin (H&E) for microscopic examination. A blinded observer assessed the severity of injury based on criteria including alveolar congestion, hemorrhage, inflammation infiltration, and alveolar wall thickness.
Wet/Dry (W/D) Weight Ratio of Lung Tissues
The W/D weight ratio of lung tissues was determined to assess pulmonary edema. Tissues were initially weighed to obtain their wet weight, then dried at 60°C for 48 hours to measure the dry weight. The ratio of wet weight to dry weight represented the W/D ratio.
Western Blot Analysis
Western blot analysis was conducted on lung tissue or MH-S cell protein extracts, with protein concentrations quantified using a BCA protein assay. Proteins were separated by SDS-PAGE, transferred to polyvinylidene difluoride (PVDF) membranes, and incubated with primary antibodies targeting NRF2, KEAP1, HO-1, and β-actin, followed by HRP-conjugated secondary antibodies. Bands were visualized using enhanced chemiluminescence and quantified via densitometry.
ELISA
ELISA quantified IL-1β, IL-6, and TNF-α levels in bronchoalveolar lavage fluid (BALF) or cell culture supernatants following the manufacturer’s protocols.
Smear the BALF
BALF was obtained by instilling and aspirating 1 mL of PBS through the trachea. Cells from the BALF were centrifuged, and the pellet was resuspended, smeared on slides, air-dried, and stained with Wright-Giemsa for differential cell count analysis under microscopy.
Flow Cytometry
Flow cytometry analyzed macrophage polarization in BALF cells stained with fluorochrome-conjugated antibodies against CD206 and CD86 for 30 minutes at 4°C in the dark, then washed, fixed, and analyzed. Data assessed the M1/M2 polarization status using flow cytometry software.
Statistical Analysis
Statistical analysis presented data as mean ± standard error of mean (SEM), with group differences evaluated by one-way analysis of variance (ANOVA) and Tukey’s post hoc test for multiple comparisons. Significance was set at p < 0.05, with analyses conducted using GraphPad Prism 9.0 software.
Results
Uridine Attenuates LPS-induced Inflammatory Responses in MH-S Macrophages
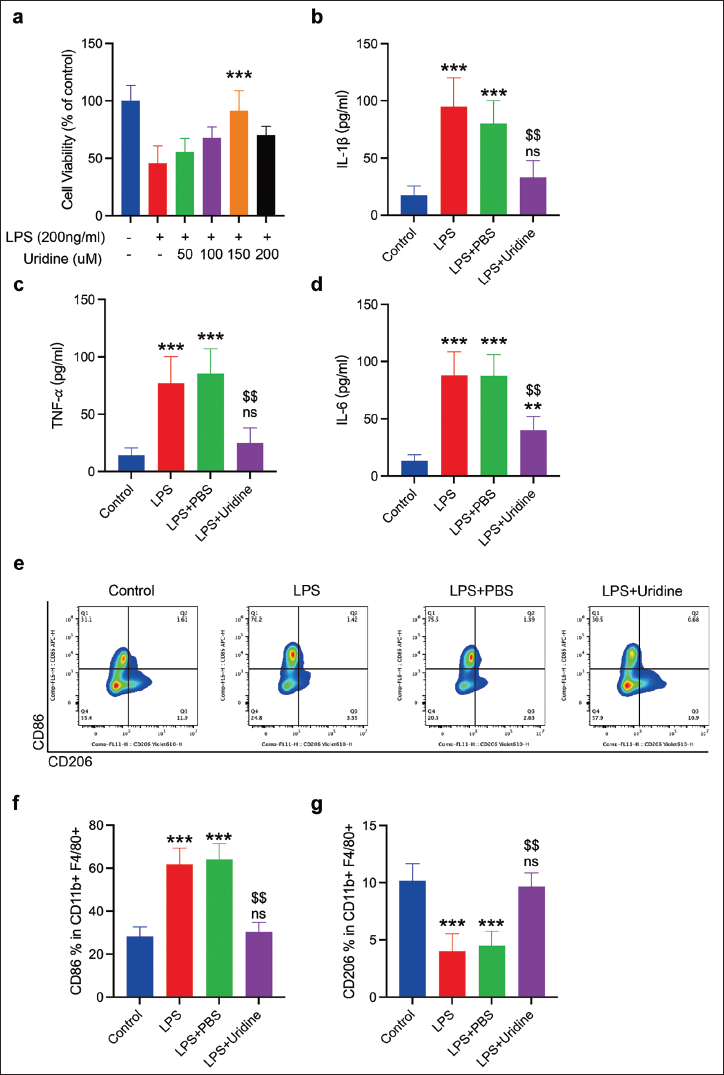
A 6-hour incubation with various concentrations of uridine assessed its cytotoxic potential through the CCK-8 assay, revealing uridine’s nontoxic nature across a 0–200 µM range. Notably, uridine enhanced the viability of MH-S macrophages impaired by LPS-induced damage, with optimal cell recovery observed at a 150 µM concentration (Figure 1a). This dose was thus identified as the most effective for further studies. LPS exposure resulted in the upregulation of pro-inflammatory cytokines IL-1β, IL-6, and TNF-α, which uridine significantly counteracted, reducing their expression (Figure 1b–d). Moreover, uridine’s impact on macrophage polarization was significant, with LPS challenge primarily inducing a pro-inflammatory M1 phenotype, marked by increased CD86+ expression. Conversely, uridine pretreatment facilitated a notable transition toward an anti-inflammatory M2 phenotype, demonstrated by the enhanced CD206+ expression within the CD11b+F4/80+ macrophage subset. This phenotypic shift underscores uridine’s capacity to modulate macrophage polarization, promoting an anti-inflammatory response to LPS stimulation (Figure 1e–g).
Uridine Mitigates Lipopolysaccharide (LPS)-induced Inflammation in MH-S Macrophages. (a) MH-S Macrophages were Incubated with Varying Concentrations (0, 50, 100, 150, and 200 µM) of Uridine for 24 Hours. Cell Viability was Assessed Utilizing the Cell Counting Kit-8 (CCK-8) Assay. Data are Presented as Mean ± SD Derived from Three Separate Experiments. Relative to the LPS Group, ***p < 0.001. (b–d) Enzyme-linked Immunosorbent Assay (ELISA) Quantified Interleukin (IL)-1β, IL-6, and Tumor Necrosis Factor-Alpha (TNF-α) Levels in the Supernatants of MH-S Cultures. (e) MH-S Macrophages were Exposed to either 200 ng/mL of LPS Alone or in Conjunction with 150 µM Uridine Pretreatment for 2 Hours, Followed by a 6-hour Treatment Period. The M1/M2 Polarization Status of Macrophages was Evaluated via Flow Cytometry. (f and g) Quantification of CD86+ (M1) and CD206+ (M2) Subsets within the CD11b+F4/80+ MH-S Macrophage Population. Data are Depicted as Mean ± SD, based on Three Independent Experiments. In Comparison to the Control Group, ***p < 0.001, ns: Not Significant; Compared to the LPS + Phosphate-buffered Saline (PBS) Group, $$p < 0.01.
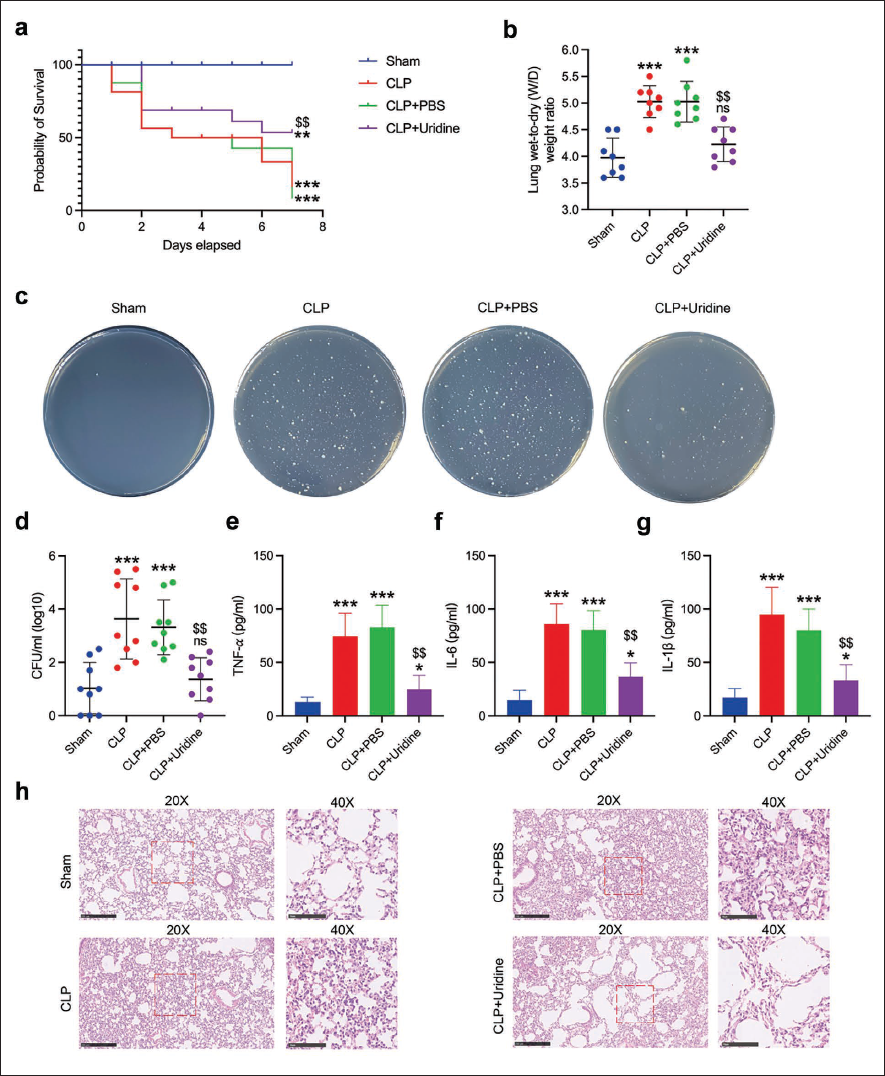
Uridine Improves Survival and Mitigates Pulmonary Damage in Septic Mice
Our exploration of uridine’s therapeutic benefits for sepsis-induced lung injury showed significant enhancements in survival rates and pulmonary health following uridine administration. Survival analysis conducted over a 7-day period post-CLP illustrated a notable improvement in the survival rate of septic mice treated with uridine compared to their untreated counterparts, underscoring uridine’s protective capacity in septic conditions (Figure 2a). Further assessments of uridine’s influence on pulmonary injury revealed a marked reduction in the lung wet-to-dry weight ratio 24 hours post-CLP, indicating diminished pulmonary edema and suggestive of enhanced pulmonary function (Figure 2b). Additionally, the antibacterial properties of uridine were evaluated by quantifying lung bacterial colonization, with findings revealing a significant decrease in bacterial load in uridine-treated mice, highlighting uridine’s role in bolstering host defenses against infection (Figure 2c and d). Pulmonary inflammatory responses were also assessed, with cytokine measurements in BALF showing substantially lower levels of IL-1β, IL-6, and TNF-α in uridine-treated mice, pointing to effective mitigation of pulmonary inflammation (Figure 2e–g). H&E staining of lung tissues after CLP treatment revealed a marked reduction in neutrophil infiltration and congestion in uridine-treated mice compared to control groups (Figure 2h), demonstrating uridine’s protective effects in preserving lung architecture and mitigating inflammatory responses. Collectively, these outcomes demonstrate uridine’s ability to improve survival, lessen pulmonary edema, reduce bacterial colonization, decrease pro-inflammatory cytokine expression in BALF, and enhance histopathological conditions in a mouse model of sepsis-induced lung injury, affirming uridine’s potential as a therapeutic agent for improving pulmonary health in sepsis.
Uridine Administration Ameliorated Pulmonary Injury in Septic Mice. The Survival Rate over a 7-day Period Post-cecal Ligation and Puncture (CLP) was Assessed with or without Uridine Treatment. (b) Pulmonary Edema was Quantified by the Lung Wet-to-dry Weight Ratio 24 Hours Following CLP. (c and d) Bacterial Colonization within the Pulmonary Tissues of Septic Mice was Evaluated. (e–g) Interleukin (IL)-1β, IL-6, and Tumor Necrosis Factor-Alpha (TNF-α) Concentrations in Bronchoalveolar Lavage Fluid (BALF) were Measured Using Enzyme-linked Immunosorbent Assay (ELISA). (h) Histopathological Examination was Conducted on Lung Sections Stained with Hematoxylin and Eosin (H&E), with a Scale Bar Representing 500 µm. Data are Represented as Mean ± SD from Three Distinct Experiments. In Comparison to the Control Group, **p < 0.01, ***p < 0.001, Not Significant (ns); Compared to the Lipopolysaccharide (LPS) + Phosphate-buffered Saline (PBS) Group, $$p < 0.01.
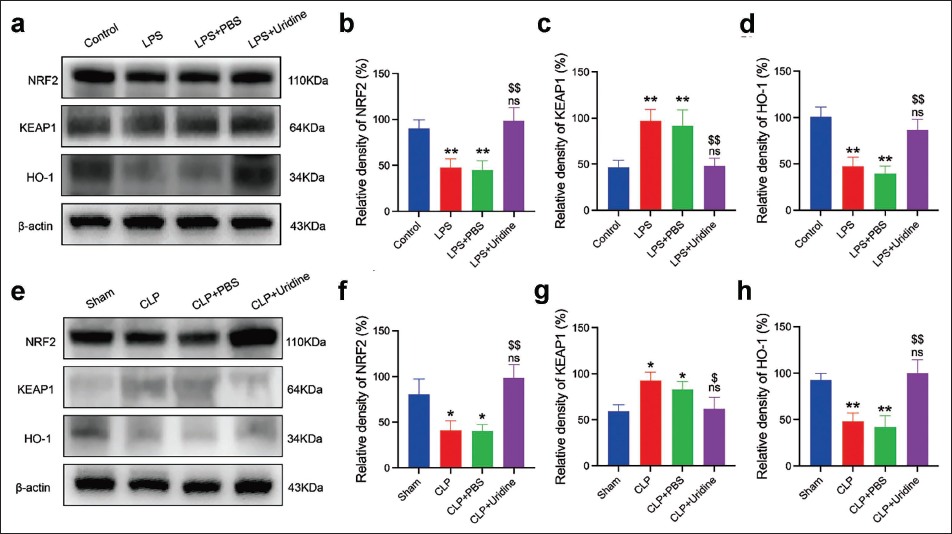
Uridine Modulates Antioxidative Response Proteins in LPS-stimulated MH-S Macrophages and Septic Mice
Western blot analysis was employed to explore uridine’s impact on antioxidative response protein expression within MH-S macrophages and septic mice. The results showcased uridine’s capacity to significantly enhance the expression of NRF2 and HO-1 proteins while concurrently reducing KEAP1 levels in MH-S cells exposed to LPS. This change signifies the NRF2 pathway’s activation, which is pivotal for the antioxidative response (Figure 3a–d). Similarly, in septic mice lung tissues, uridine administration markedly elevated NRF2 and HO-1 levels and decreased KEAP1 expression, reinforcing uridine’s role in amplifying antioxidative defense mechanisms (Figure 3e–g). These outcomes indicate uridine’s protective efficacy against LPS-induced oxidative stress and inflammation is partially mediated through the modulation of the NRF2 signaling pathway. The simultaneous upregulation of NRF2 and HO-1, along with KEAP1 downregulation, emphasizes uridine’s capacity to strengthen cellular defenses against oxidative harm in both in vitro and in vivo sepsis models.
Uridine-induced Activation in Lipopolysaccharide (LPS)-stimulated MH-S Macrophages and Septic Mice. (a) Western Blot Analyses were Performed to Assess the Expression Levels of NRF2, KEAP1, and HO-1 Proteins in MH-S Cells across Various Treatment Groups. (b–d) Quantitative Evaluation of NRF2, KEAP1, and HO-1 Protein Levels in the Specified Groups. (e) Protein Expression of NRF2, KEAP1, and HO-1 in the Lung Tissues of Septic Mice was Determined via Western Blotting for the Indicated Groups. (f–g) Comparative Analysis of NRF2, KEAP1, and HO-1 Levels in the Lung Tissues of Septic Mice among the Designated Groups. Data are Presented as Mean ± SD, based on Results from Three Independent Experiments. When Compared to the Control Group, *p < 0.05, *p < 0.01, ns: Not Significant (ns); Compared to the LPS + Phosphate-buffered Saline (PBS) Group, $p < 0.05, $$p < 0.01.
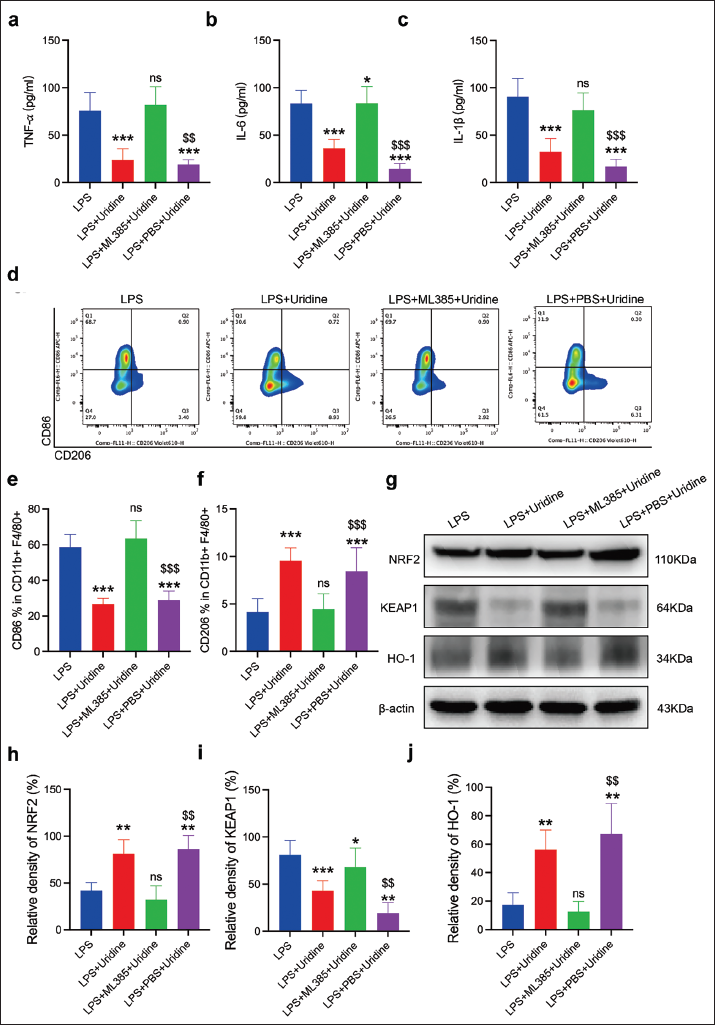
Effect of ML385 on Uridine’s Anti-inflammatory Actions in LPS-stimulated MH-S Macrophages
The introduction of ML385 prior to uridine treatment resulted in a notable surge in pro-inflammatory cytokine levels (IL-1β, IL-6, and TNF-α) in cell supernatants, as determined by ELISA (Figure 4a–c). This indicates ML385’s efficacy in counterbalancing uridine’s cytokine-suppressing properties, highlighting the NRF2 pathway’s crucial role in uridine-driven anti-inflammatory responses. Flow cytometry analysis further revealed ML385’s capacity to diminish the uridine-induced shift toward an M2 macrophage phenotype, evidenced by altered proportions of CD86+ (M1) and CD206+ (M2) cells within the CD11b+F4/80+ MH-S macrophage ensemble (Figure 4d–f). This observation accentuates the significance of NRF2 activation in influencing macrophage polarization and underscores ML385’s ability to negate uridine’s effects on macrophage phenotypic modulation. Subsequent Western blot analysis and quantitative assessments of NRF2, KEAP1, and HO-1 protein expressions confirmed ML385’s role in reversing the expression patterns induced by uridine (Figure 4g–j). ML385’s action thus crucially implicates the NRF2 signaling pathway in orchestrating the anti-inflammatory and antioxidative responses elicited by uridine in LPS-challenged MH-S macrophages.
ML385, a Specific NRF2 Inhibitor, Reverses the Anti-inflammatory Effects of Uridine in MH-S Macrophages Stimulated with Lipopolysaccharide (LPS). (a–c) Interleukin (IL)-1β, IL-6, and Tumor Necrosis Factor-Alpha (TNF-α) Concentrations in the Supernatants of MH-S Cells were Quantified Using Enzyme-linked Immunosorbent Assay (ELISA). (d) MH-S cells Underwent Treatment in the Presence or Absence of 200 ng/mL of LPS for 6 hours, with or without a 2-hour Pretreatment of Uridine (150 µM). The M1/M2 Macrophage Polarization was Assessed through Flow Cytometry. (e and f) Proportions of CD86+ (M1) and CD206+ (M2) Phenotypes within the CD11b+F4/80+ MH-S Macrophage Population were Determined. (g) Western Blot Analysis was Conducted to Evaluate NRF2, KEAP1, and HO-1 Protein Expression in MH-S Cells among the Specified Groups. (h–j) Quantitative Analysis of NRF2, KEAP1, and HO-1 Protein Levels in the Designated Groups. Data are Reported as Mean ± SD, Stemming from Three Separate Experiments. Relative to the Control Group, *p < 0.05,**p < 0.01,***p < 0.001, Not Significant (ns); Compared to LPS + Phosphate-buffered Saline (PBS) Group, $$p < 0.01, $$$p < 0.001.
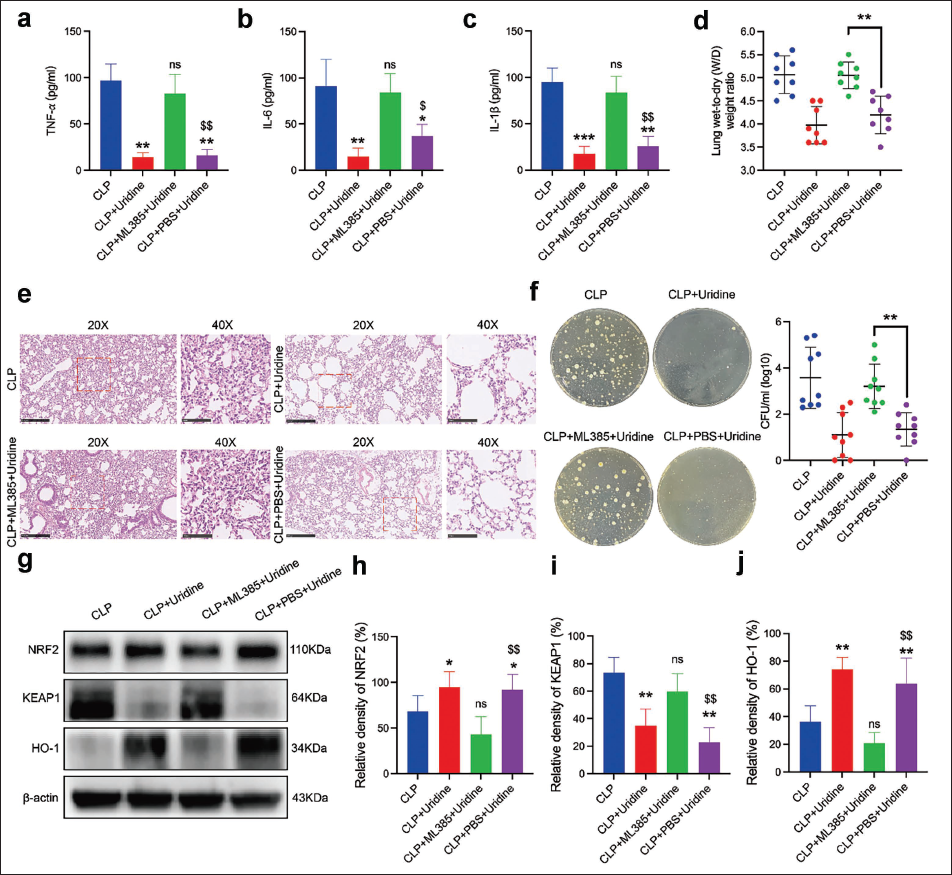
Impact of ML385 on the Protective Effects of Uridine in Septic Mice
Pre-administering ML385 before uridine led to elevated pro-inflammatory cytokine levels (IL-1β, IL-6, and TNF-α) in the BALF, effectively counteracting uridine’s anti-inflammatory advantages (Figure 5a–c). This suggests ML385’s NRF2 inhibition directly challenges uridine’s ability to suppress cytokine secretion in septic mice’s lungs. Moreover, the lung wet-to-dry weight ratio, indicative of pulmonary edema, was higher in the group treated with both ML385 and uridine compared to the uridine-only group, signaling increased pulmonary edema and reduced uridine efficacy in mitigating lung tissue fluid retention (Figure 5d). Histological observations further revealed intensified inflammatory infiltration and tissue damage in mice receiving both ML385 and uridine, compared to those treated with uridine alone, reinforcing the idea that ML385 negates uridine’s lung protection benefits (Figure 5e). Bacterial burden quantification in the lungs showed ML385 counteracted the improved bacterial clearance achieved with uridine, resulting in higher bacterial loads (Figure 5f). Western blot analysis of lung tissues underscored ML385’s reversal of uridine’s modulatory effects on NRF2, KEAP1, and HO-1 levels, emphasizing the NRF2 pathway’s critical role in uridine’s protective mechanism against septic lung injury (Figure 5g–j).
ML385 Abrogated the Beneficial Effect of Uridine on Mitigating Acute Lung Injury in Septic Mice. (a–c) Concentrations of Interleukin (IL)-1β, IL-6, and Tumor Necrosis Factor-Alpha (TNF-α) in Bronchoalveolar Lavage Fluid (BALF) were Determined Using Enzyme-linked Immunosorbent Assay (ELISA). (d) The Lung Wet-to-dry Weight Ratio, an Indicator of Pulmonary Edema, was Measured 24 hours Post-cecal Ligation and Puncture (CLP). (e) Histological Analysis was Performed on Lung Sections Stained with Hematoxylin and Eosin (H&E), with a Scale Bar Equivalent to 500 µm. (f) The Bacterial Burden within the Lungs of Septic Mice was Quantified. (g) Western Blot Analysis Assessed the Expression Levels of NRF2, KEAP1, and HO-1 Proteins in the Lung Tissues of Septic Mice across Various Treatment Groups. (h–j) Quantitative Evaluation of NRF2, KEAP1, and HO-1 Protein Levels in the Lung Tissues of Septic Mice was Conducted among the Specified Groups. Data are Presented as Mean ± SD, Derived from Three Independent Experiments. Compared to Control Group, *p < 0.05,**p < 0.01,***p < 0.001, ns: Not Significant; Compared to Lipopolysaccharide (LPS) + Phosphate-buffered Saline (PBS) Group, $p < 0.05, $$p < 0.01.
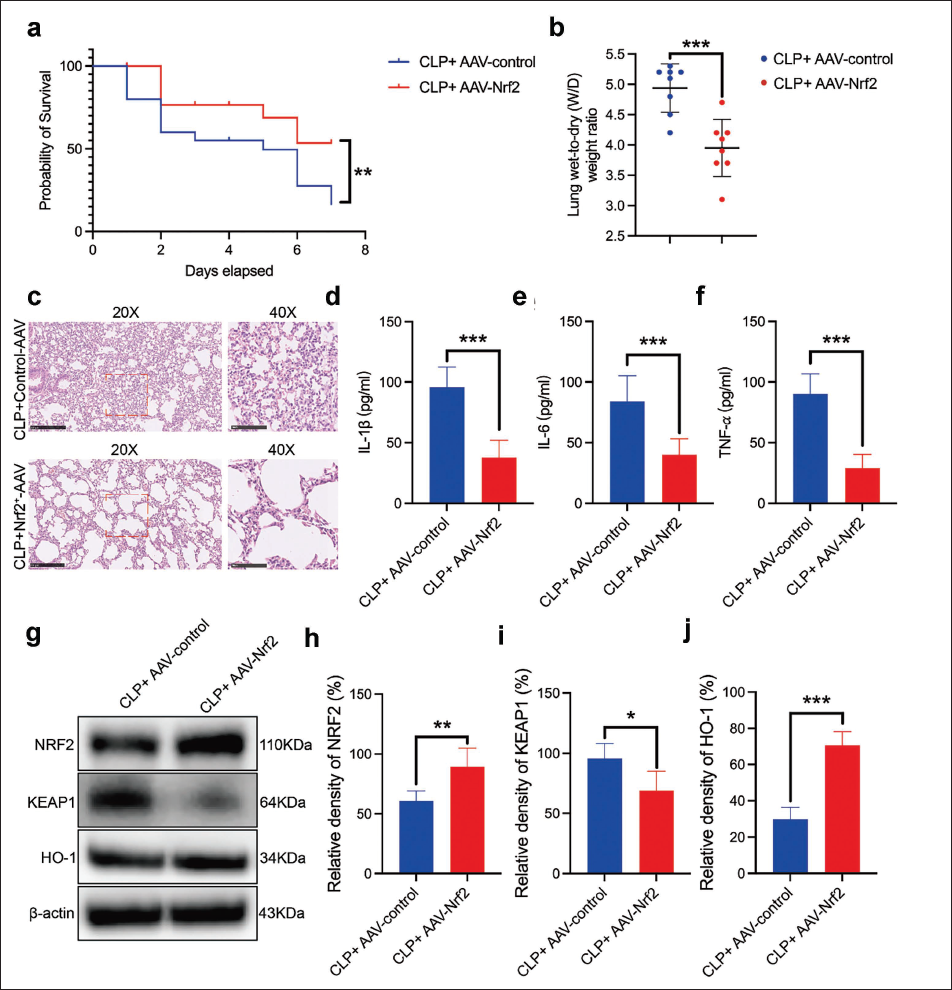
Efficacy of Nrf2 Overexpression in Ameliorating Sepsis-induced ALI
Utilizing an adeno-associated virus (AAV) engineered for Nrf2 overexpression, we administered it intratracheally to mice 3 weeks before the CLP procedure to upregulate NRF2 in the lungs. Mice with enhanced NRF2 expression demonstrated markedly improved survival rates following CLP, indicating the potential protective benefits of targeted Nrf2 overexpression in mitigating the impact of sepsis (Figure 6a), alongside a notable decrease in pulmonary edema as shown by lower lung wet-to-dry weight ratios, suggesting less fluid accumulation and possibly enhanced lung function (Figure 6b). H&E of lung sections confirmed Nrf2 overexpression’s protective effect, displaying lesser inflammatory infiltration and better-maintained lung structure than the control (Figure 6c). The reduction of pro-inflammatory cytokines IL-1β, IL-6, and TNF-α in BALF accentuated the anti-inflammatory benefits of Nrf2 overexpression, highlighting its pivotal role in reducing pulmonary inflammation (Figure 6d–f). Western blot analysis further demonstrated significant upregulation of NRF2 and HO-1, alongside KEAP1 downregulation in the lungs of Nrf2-overexpressing septic mice (Figure 6g–j), verifying the activation of anti-inflammatory and antioxidative pathways through Nrf2 modulation, thereby reinforcing its therapeutic potential in treating sepsis-induced lung injury.
Nuclear Factor Erythroid 2-related Factor 2 (Nrf2) Overexpression Mitigated Acute Lung Injury following Sepsis. (a) The 7-day Survival Rate Post-cecal Ligation and Puncture (CLP) was Evaluated with or without Uridine Treatment. (b) Pulmonary Edema was Quantified via the Lung Wet-to-dry Weight Ratio 24 Hours after CLP. (c) Lung Sections were Histologically Examined and Stained with Hematoxylin and Eosin (H&E), with a Scale Bar of 500 µm. (d–f) Levels of Interleukin(IL)-1β, IL-6, and Tumor Necrosis Factor-Alpha (TNF-α) in Bronchoalveolar Lavage Fluid (BALF) were Measured Using Enzyme-linked Immunosorbent Assay (ELISA). (g) Expression Levels of NRF2, KEAP1, and HO-1 Proteins in the Lung Tissues of Septic Mice were Assessed through Western Blot Analysis in the Indicated Groups. (h–j) Quantitative Evaluation of NRF2, KEAP1, and HO-1 Protein Levels in the Lung Tissues of Septic Mice was Performed among the Specified Groups. Data are Represented as Mean ± SD, based on Three Separate Experiments. Significance Levels are Denoted as *p < 0.05, **p < 0.01,***p < 0.001.
Discussion
This investigation elucidates uridine’s potential in alleviating sepsis-induced pulmonary damage by activating the Keap1-Nrf2 signaling pathway. It highlights uridine’s multifaceted therapeutic benefits, including its ability to modulate macrophage polarization toward an anti-inflammatory state, bolster antioxidative defenses, and significantly improve survival rates. Uridine protects against sepsis-induced lung damage, as evidenced by the decreased pulmonary bacterial colonization and reduced lung wet-to-dry weight ratio in uridine-treated septic mice.
Given that sepsis is a major cause of death in critical care units around the globe, the noteworthy improvement in survival rates with uridine administration is very noteworthy (Jiang et al., 2019). This result is consistent with other research emphasizing the cytoprotective and anti-inflammatory properties of nucleosides in systemic inflammatory settings (Lai et al., 2023). Due to its broad effects on immunological regulation, reduction of oxidative stress, and correction of metabolic disturbances—all important components in the pathophysiology of sepsis—uridine is thought to contribute to higher survival rates (Lairion et al., 2023; Mironova et al., 2018).
Histopathological evaluations show that uridine reduces lung damage, which is consistent with uridine’s well-known anti-inflammatory characteristics. This capacity most likely results from uridine’s ability to inhibit the generation of cytokines that promote inflammation, a feature that has been shown in several different inflammation models (Jeengar et al., 2017; Luo et al., 2021). Furthermore, the significant reduction in lung bacterial burden raises the possibility of improving the innate immune system, maybe via upregulating antimicrobial peptides, which calls for additional research.
Furthermore, the notable decrease in lung wet-to-dry weight ratios seen in mice treated with uridine highlights the drug’s efficacy in reducing pulmonary edema, which plays a critical role in the emergence of ALI and ARDS, which are common post-sepsis outcomes. The abnormal build-up of fluid in lung tissues, known as pulmonary edema, seriously hinders gas exchange and increases the risk of respiratory failure (Murray, 2011). In addition to demonstrating uridine’s ability to preserve lung structure and function, its ability to reduce fluid build-up suggests that uridine may play a role in maintaining vascular integrity and reducing capillary leakage, possibly by controlling inflammatory mediators that impair endothelial barrier function.
The body uses Nrf2 as a key component in its defense against oxidative stress (Cuadrado et al., 2018; Li et al., 2019). In addition to directly suppressing the transcription of pro-inflammatory genes like IL-1β and IL-6, it subtly reduces the function of nuclear factor kappa B (NF-κB), an essential regulator of inflammation. Our findings highlight the critical function of the Keap1-Nrf2 signaling pathway in augmenting uridine’s protective effect against lung damage. Inflammation and oxidative stress play important roles in the pathophysiology of ALI/ARDS (Fan et al., 2018; Liang et al., 2019). One important way uridine provides protection is by activating Nrf2, which leads to the overexpression of target antioxidative genes like HO-1. This is consistent with previous research that shows the Keap1-Nrf2 axis to be a key component of cellular defense systems against oxidative stress (Gümüş et al., 2022; Ma, 2013; Wang et al., 2020). Additionally, the use of ML385, a known inhibitor of the Nrf2 pathway, effectively counteracted the beneficial effects of uridine, demonstrating the crucial role that this signaling pathway plays in enabling uridine’s protective benefits. This reversal supports the underlying mechanism of uridine’s therapeutic potential and emphasizes the potential benefit of focusing on the Nrf2 pathway as a fresh approach to sepsis therapy development.
Although our findings are positive, they also emphasize the need for a deeper understanding of the complex relationships between uridine’s immunomodulatory, antibacterial, and antioxidant properties. Furthermore, it is crucial to identify the precise methods by which uridine triggers the Keap1-Nrf2 pathway and its ensuing functions in regulating immunity and aiding in removing microorganisms. Subsequent investigations must focus on pinpointing the exact molecular mechanisms via which uridine influences macrophage activity, enhances bacterial clearance, and fortifies the antioxidative defense system. Furthermore, translating these preclinical results into clinical settings will need a sophisticated comprehension of the multifaceted nature of sepsis and the different variables influencing patient outcomes.
Conclusion
In conclusion, this study demonstrates the therapeutic potential of uridine in treating pulmonary damage brought on by sepsis by activating the Keap1-Nrf2 pathway. These findings provide light on the protective mechanisms of uridine and also establish the foundation for future investigations into its potential therapeutic value for sepsis and other inflammatory diseases. Subsequent studies must focus on deciphering the complex molecular mechanisms behind uridine’s effects and evaluating its therapeutic potential in real-world settings.
Footnotes
Abbreviations
ALI: Acute lung injury; ARDS: Acute respiratory distress syndrome; AAV: Adeno-associated virus; BALF: Bronchoalveolar lavage fluid; CCK-8: Cell Counting Kit-8; CLP: Cecal ligation and puncture; ELISA: Enzyme-linked immunosorbent assay; FBS: Fetal bovine serum; H&E: Hematoxylin and eosin; HO-1: Heme oxygenase-1; HRP: Horseradish peroxidase; IL: Interleukin; Keap1: Kelch-like ECH-associated protein 1; LPS: Lipopolysaccharide; Nrf2: Nuclear factor erythroid 2-related factor 2; TNF: Tumor necrosis factor; W/D: Wet/dry.
Acknowledgments
The authors thank Yangtze University for providing the research platform.
Authors’ Contributions
Data curation, Guoping Li and Yalan Hu; Formal analysis, Guoping Li; Funding acquisition, Fei Li; Investigation, Guoping Li and Yalan Hu; Methodology, Guoping Li, Yalan Hu, Fan Xu, and Fei Li; Project administration, Fei Li; Resources, Fei Li; Software, Guoping Li and Fan Xu; Supervision, Fei Li; Visualization, Fei Li; Writing – original draft, Guoping Li and Yalan Hu; Writing – review & editing, Fei Li.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Ethical Approval and Informed Consent
NA.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the China Association for Promotion of Health Science and Technology (JKH2023015-13).