
Abstract
Introduction
Cannabidiol (CBD) is one of many naturally biosynthesized compounds produced by Cannabis sativa. There is limited information available in the literature on hydrogenated CBD (tetrahydro cannabidiol or H4CBD) (Adams et al., 1940b). As hydrogenated derivatives of tetrahydrocannabinol (THC) and CBD become increasingly popular in consumer markets, toxicological assessments are vital in identifying toxic characteristics, if any, caused by hydrogenated cannabinoids.
Objectives
Assessment of the preclinical toxicology of hydrogenated CBD is provided through the in vitro safety study of racemic H4CBD in hepatocytes, normal human lung fibroblasts (NHLF), and primary human neural progenitor (NPC) cell lines. The importance of these cell lines is related to major organs and is the primary focus in determining any major toxic characteristics when consuming products. The inclusion of the human ether-a-go-go related gene (hERG) patch clamp test, observes any inhibition of sodium and potassium ion channels related to the arrhythmia of the heart. Also, the AMES test was conducted to determine any carcinogenic characteristics that H4CBD might impose.
Materials and Methods
Plated NHLF, hepatocytes, and NPC were used in a preclinical 3-[4,5-dimethylthiazol-2-yl]-2,5 diphenyl tetrazolium bromide (MTT) assay for cytotoxicity observations with the visible color change of cellular use of formazan, while a plated AMES test was conducted to monitor any visible mutations within Escherichia coli for carcinogenic activity. Plated cloned HEK293 cells were given set voltages to determine ion channel activity to determine if H4CBD causes inhibition within these pathways, which would mimic any arrhythmia potential in cardiomyocytes.
Results
Screening of the MTT assay had a median calculated 3.25 micromolar concentration where cell viability remained high in NHLF and NPC, with higher concentrations leading to decreased cell viability. A 3.25 micromolar concentration is also the median for hepatocytes, with a discrepancy in some of the data that could be accounted for by miscounting colonies. The hERG patch clamp test provided a zero net inhibition with values adding up to zero, determining that the compound did not inhibit normal processes within the ion channels of the plated HEK293 cells. The analysis of the different cell types revealed varying responses to H4CBD. NHLF exhibited a concentration-dependent reduction in cell viability, with sustained concentrations over 24 h at 6.25 µM resulting in a significant loss of viability. Conversely, hepatocytes showed a trend of decreased viability at longer exposure times and higher concentrations, but severe cytotoxicity was not observed. This suggests that hepatocytes are less susceptible to the cytotoxic effects of H4CBD compared to NHLF. In the hERG assay, H4CBD did not inhibit the action potentials within cardiomyocytes, indicating no inhibition of ion channels involved in cardiac function. This finding is important for assessing the potential cardiovascular effects of H4CBD. The AMES test results were negative, indicating that H4CBD did not demonstrate mutagenic activity in the tested strains.
Conclusion
This supports the conclusion that H4CBD does not possess carcinogenic potential based on this assay. Both human NPC and NHLF showed a median concentration of 3.25 µM before exhibiting a significant reduction in cell viability. This information is valuable for determining research or consumer usage limits for H4CBD. It is important to note that this study represents a preclinical assessment, and further studies are required. The experimental design followed protocols typically used for preclinical assessments of novel pharmaceuticals. These findings provide insight into the cytotoxic effects of H4CBD and contribute to establishing research and safety parameters as these compounds continue to gain attention.
Introduction
Cannabis has been around for thousands of years and is even used in medicine within the United States, freely mixed in tonics and various other mixtures described in the United States Pharmacopeia in 1850 (Jacob & Todd, 1940). Due to legal ramifications and political duress, cannabis was dropped from the United States Pharmacopoeia in the 1940s and labeled as a controlled substance in the 1970s. The fluctuation in favor of cannabis has limited advancements in the field of cannabinoid chemistry (Bridgeman & Abazia, 2017).
The first cannabinoid(s) was not elucidated until the early 1940s, when cannabinol (CBN) was identified, following cannabidiol (CBD), which was identified in 1942. As research advances with cannabinoids, rare cannabinoids that have been recently elucidated are garnering attention and popularity, while knowledge of these cannabinoids and their effects remains limited or nonexistent. CBD is one of the hundreds of currently identified cannabinoids that are biosynthesized by Cannabis sativa naturally in various concentrations (Adams et al. 1940a, b), including but not limited to the C. indica or C. ruderalis subspecies (Welty et al., 2014). A multitude of studies have been performed on CBD and tetrahydrocannabinol (THC), evaluating their safety and effects on certain ailments, including inflammation and antiproliferative/proapoptotic effects within the body (Seltzer et al., 2020).
Of the limited studies produced on cannabinoid derivatives, minute amounts of data are produced on their hydrogenated cannabinoid derivatives (Appendino, 2020; Bloom et al., 1977). Several studies that have been published have focused on the synthetic hydroxyl derivatives of THC, such as 9-nor-9β-hydroxyhexahydrocannabinol (9-nor-9β-HHC), 9-hydroxyhexahydrocannabinol (9-OH-HHC), or 11-hydroxyhexahydrocannabinol (11-OH-HHC and 7-OH- HHC), commonly confused with hexahydrocannabinol (HHC), the hydrogenated scaffold of THC, in research and consumer markets; due to the nomenclature used, none has focused on the hydrogenated derivatives of CBD, such as tetrahydro cannabidiol (H4CBD).
As the popularity of cannabinoids skyrockets, so does the need for markets to continually update with derivatives that are homologous to THC and CBD to meet the demands of the cannabinoid market. These new/rediscovered cannabinoids have no preclinical safety profile performed on them and are being consumed. We performed a preclinical assessment on the cyclic cannabinoid HHC, performing experiments designed for novel pharmaceuticals to provide a preclinical assessment profile for consumption and other studies to be performed (Collins et al., 2023). Since the discovery of H4CBD in 1940 through the catalytic hydrogenation of CBD, H4CBD has been manufactured at a large scale to meet market demand. The rediscovery of these hydrogenated derivatives is making its way into the medicinal chemistry. Comparing the similar pharmacophores that they share with their parental counterparts (CBD and THC), they could also be used for the treatment of ailments and diseases. In an earlier study produced by Ben-Shabat et al., hydrogenated cannabinoid derivatives of CBD and the dimethyl heptyl-CBD scaffolds, which included a mixture of H4CBD diastereomers, demonstrated that the diastereomers of H4CBD bound to the CB1 receptor with great affinity (145 nM), as well as their potential to be used as anti-inflammatory and anti-cancer agents (Ben-Shabat et al., 2006). There are some preliminary studies on the mechanism and the binding affinities of H4CBD that have been produced, but unfortunately, there are no safety studies on H4CBD.
Unlike CBD, H4CBD has been reported as an agonist of the CB1 receptor. There is no deep structural characterization of H4CBD, and we sought to contribute to the field by contributing a full characterization of HHC and H4CBD (Collins et al., 2022, 2023). Furthermore, the lack of any preclinical assessment data leaves gaps for use in research as information on cytotoxicity, genotoxicity, and possible repolarization of ion channels for H4CBD is absent. Consumption of H4CBD is increasing due to the declared physiological effects of desired relaxation and anti-inflammatory properties claimed by users while also claiming slight intoxication, with intoxication lasting for short amounts of time. These claims are increasing the popularity and usage of H4CBD, with due diligence needed to provide an assessment of sorts. Our contribution includes several experiments at the preclinical level that were designed to enhance and provide an in-depth assessment of the safety of a mixture of H4CBD diastereomers. Cultured human neural progenitor cells (NPC) (Martínez-Cerdeño & Noctor, 2018), cultured normal human lung fibroblasts (NHLF) (Roberts et al., 2021), and primary human hepatocytes (PHH) (Schulze et al., 2019) were used as specific targets for evaluation at various concentrations of H4CBD to determine in vitro cytotoxic effects. The cardiac safety profile was generated using a common test, the human ether-a-go-go related gene (hERG) patch clamp test (Sanguinetti & Tristani-Firouzi, 2006), used in preclinical pharmaceutical assessments for identifying any repolarization issues of ion channels within the cardiomyocytes where the repolarization of the current in the cardiac action potential can lead to fatal arrhythmias (i.e., Torsades de Pointes). An AMES test (Föllmann et al., 2013) was conducted as well to identify any genotoxic effects that H4CBD might produce at varying concentrations. These experiments were designed with the testing of H4CBD following most preclinical assessments of pharmaceuticals to determine the safety of the compound (Dziwenka & Coppock, 2022; Ladin et al., 2016).
Materials and Methods
Chemicals, Reagents, and Instruments
CBD was used as a starting material for the synthesis of H4CBD. CBD was purchased from GVB Biopharma and converted following literature procedures (Gong et al., 2020; Petrzilka et al., 1967; Scialdone, 2019). Purification of the completed reaction crude afforded the desired product. H4CBD was dissolved in acetonitrile-d3 (CD3CN), and 1H, 13C, correlation spectroscopy (COSY), heteronuclear single quantum coherence (HSQC), heteronuclear multiple bond correlation (HMBC), and nuclear overhauser effect spectroscopy (NOESY) data were acquired on a 500 MHz Bruker AVANCE II system at 25°C. 1H, 13C, COSY, HSQC, and HMBC data sets were analyzed to yield complete 1H and 13C peak assignments. Once the peaks were assigned, NOESY data were analyzed to yield stereochemistry information about the orientation of the hexanyl-methyl group. Instruments used were the Shimadzu TQ8050 NX Mass Spectrometer, Shimadzu Nexis GC-2030 Gas Chromatograph, Shimadzu AOC-20i Plus Autosampler, and Agilent 1100 series high-performance liquid chromatography (HPLC) with Diode Array Detector to determine isomers using the C18-RP column. Solvents were purchased from Sigma Aldrich (Burlington, MA). Deuterated solvents were purchased from Cambridge Isotope Laboratories (Andover, MA).
Synthesis of H4CBD
A 20-L flask equipped with a reflux condenser and an addition funnel was purged with argon for 60 min at 1 bar. Ten percent palladium on carbon (Pd/C) (0.1 mol%) was slowly added to the reaction using a powder funnel under argon. The flask was then purged with argon for 60 min at 1 bar. Ethanol (10 L) was added slowly to avoid sparking the solvent. CBD (100 g) was dissolved in minimal amounts of ethanol. The solution was added to the flask under argon and purged for 60 min at 1 bar. Afterward, the atmosphere (ATM) of argon was stopped, and an ATM of hydrogen (1 bar) was introduced. The reaction was then stirred at 25°C for 72 h or until complete by HPLC. Upon completion, the reaction was purged with argon for 10 min at 1 bar. The reaction mixture was poured over filter paper (1–3 µm) on a Buchner funnel and then concentrated in vacuo. The crude oil was then dissolved in hexane and purified over silica gel (0%–5% ethyl acetate). The fractions of interest were concentrated in vacuo and then distilled to afford an orange oil, a mixture of 69.8% (R) and 29.5% (S) diastereomers of H4CBD. HPLC (C18): 4.610, 4.697 min, 1H nuclear magnetic resonance (NMR) (500 MHz, CD3CN): δ 6.25 (2H), 6.21 (1H), 6.19 (1H), 5.42 (1H), 3.10 (1H), 2.48 (3H), 2.25 (1H), 1.84 (1H), 1.62 (4H), 1.54 (3H), 1.4 (6H), 1.15 (2H), 0.96 (7H), 0.91 (4H), 0.78 (4H). In the case of H4CBD isomers, a pseudomolecular ion at m/z 318 based on positive ionization electrospray liquid chromatography–mass spectrometry (LC-MS) analysis indicated an apparent molecular weight of 318, consistent with that of the H4CBD isomers. Analysis of the sample by gas chromatography/mass spectrometry indicated the presence of a molecular mass of 318 and was characterized by fragment ions at m/z 193, m/z 233, and m/z 262.
Synthesis, Purification, and Characterization of H4CBD
Partial hydrogenation of (−)-CBD was carried out using 10% Pd/C as a catalyst at 1 bar for 4 h. This procedure yields a mixture of H2−CBD (1 and 1a). Full hydrogenation was completed in 72 h and led to the tetrahydro-CBD creating a stereogenic center at the C-5 position (Scheme 1). Two epimers, 9R and 9S, were generated with 69.8% and 29.5%, respectively (Collins et al. 2023). These diastereomers of H4CBD were separated by supercritical fluid chromatography (SFC) using a chiral column, obtaining a purity higher than 99% for each one. The structural elucidation of the diastereomers was accomplished using 1H, 13C, COSY, HSQC, HMBC, and NOESY.
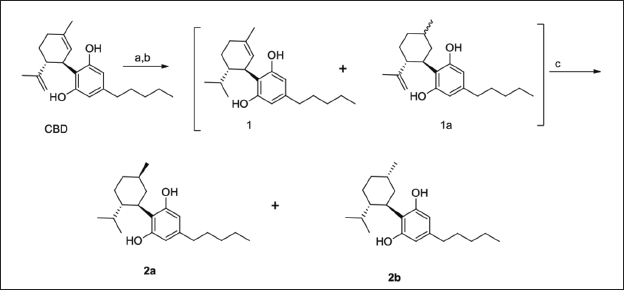
Once the peaks were assigned, NOESY data were used to confirm the orientation of the methyl group related to the hexanyl ring and determine the structure of H4CBD-2a and H4CBD-2b. In H4CBD-2a, the proton H4 shows NOE to the proton H6, but no NOE to methyl protons. This indicates that they are pointing in opposite directions (Collins et al., 2023). However, H4CBD-2b has NOE for methyl protons at 11, indicating that both are oriented in the same direction. Therefore, the structure proposed for 2b is ((1R,2S,5S)-2-isopropyl-5-methylcyclohexyl)-5-pentylbenzene-1,3-diol (Collins et al., 2023).
Chiral Separation of (R) and (S) H4CBD
The diastereomers of H4CBD were separated by SFC. SFC is used in the industry primarily for the separation of chiral molecules and uses the same columns as standard HPLC systems and CO2 as the mobile phase. The analytical and preparative SFC methods described below were used for the purification. Testing was not conducted on specific diastereomeric isolates, as the general periclinal testing of this product was to provide insight into marketed products in which isolated diastereomers can be bought but are primarily sold as a diastereomeric mixture.
Analytical SFC Method
Column 4.6 × 100 mm Chiralpak AD-H from Chiral Technologies (West Chester, PA); CO2 co-solvent (Solvent B) isopropanol; isocratic method 25% co-solvent at 4 mL/min system pressure 125 bar; column temperature 40°C; and sample diluent isopropanol.
Preparative SFC Method
Column 2.1 × 25.0 cm Chiralpak AD-H from Chiral Technologies (West Chester, PA); CO2 co-solvent (Solvent B) isopropanol; isocratic method 30% co-solvent at 80 g/min; system pressure 100 bar; column temperature 25°C; and sample diluent isopropanol.
The H4CBD was purified using the preparative method described above. The collected fractions were dried in a rotary evaporator at 40°C and transferred to the final container using isopropanol. The isolates were analyzed, and recovery details for each isolate are outlined in Table 1. The percentage of each isomer was calculated by SFC (UV220 nM). The chromatograms of the crude mixture and the isolated products are shown in Supporting Information.
IC50 Value of H4CBD and Mean Percentages of hERG Inhibition.
For the NMR analysis, the samples of (R) and (S) H4CBD were dissolved in CD3CN, and 1H, 13C, COSY, HSQC, HMBC, and NOESY data were acquired on a 500 MHz Bruker AVANCE II system at 25°C. In order to avoid the replacement of hydroxyl protons with deuterium and an easier recovery of the product from CD3CN from the NMR sample than when dissolved in dimethyl sulfoxide-d6 (DMSO-d6), deuterated acetonitrile (MeCN) was preferred over other NMR solvents. 1H, 13C, COSY, HSQC, and HMBC data sets were analyzed to yield complete 1H and 13C peak assignments. The data is provided in the supporting information.
Control Items for NHLF, NPC, and PHH
Negative Control
The negative control is 1% DMSO in the assay media. The negative control is culture media with 1% DMSO.
Positive Control
The positive control is 1% sodium dodecyl sulfate (SDS) in the assay media. Ten percent SDS was prepared in sterile water (10 mg in 1 mL). This stock was diluted in the assay medium to 1%.
Test Article Dose Formulation
Six concentrations of H4CBD were prepared. The final concentrations were 50, 25, 12.5, 6.25, 3.13, and 1.56 µM. An initial 5 mM stock (100×) was made by dissolving ~1.6 mg H4CBD per 1 mL DMSO. From the initial 5 mM (100× of the first target concentration), serial dilutions of 1:1 in DMSO were performed to obtain the 100× stock for the other target concentrations. Homogeneity, concentration, and stability analyses were not performed.
Test System
NHLFs are isolated from adult lung tissue. The PHH sourced for this study were pooled from five individuals and selected to be plateable. The NPCs used in this study are derived from induced human pluripotent stem cells.
Cell Culture
Cryopreserved NHLFs (Lonza) were thawed and grown in the fibroblast cell basal medium plus fibroblast cell medium bullet kit according to CRL-CLE SOPs. Cells were plated at a concentration of 10,000 cells/well in 96-well plates and incubated overnight before exposure.
Cryopreserved NPCs (ATCC) were thawed and grown in DMEM:F12 supplemented with a Growth Kit for NPC Expansion and 1% pen/strep according to CRL-CLE SOPs. Cells were then plated at 25,000 cells/well in 96-well plates coated with CellMatrix Basement Membrane Gel (ATCC) and incubated overnight prior to exposure.
Pooled plateable human cryopreserved hepatocytes (from five different donors) were thawed, deposited in Hepatocyte Thaw Medium (Gibco), spun down, resuspended, and plated directly into 96-well plates with Williams E media supplemented with Primary Hepatocyte Plating Supplements (Gibco). Cells were plated at concentrations of 40,000 cells/well, cultured for 6 h, and tested at ~95% confluency. All incubations were carried out in a humid ATM containing ~5% CO2 in the air in the dark at 37.0 ± 2.0 C. The temperature was continuously monitored throughout the experiment. The humidity was checked once on each working day per CRL-CLE SOPs.
Control and Test Article Exposures
Test article exposure media was prepared from 100× stocks (10 µL of test article per 990 µL media). Vehicle control exposure media was prepared from 10 µL of DMSO per 990 µL media. Positive control exposure media (1% SDS in media) were made by mixing 100 µL of 10% SDS with 900 µL of assay media. Growth media will be removed from cells, and wells will be refilled with test media (100 µL). Three time points were tested per cell line: 24, 48, and 72 h. At least four technical replicates were performed for all concentrations, controls, and blanks.
Viability Assessment
At the conclusion of each exposure, cells were assessed for viability by a 3-[4,5-dimethylthiazol-2-yl]-2,5 diphenyl tetrazolium bromide (MTT) assay (Sigma-Aldrich, St. Louis, MO). Briefly, after the culture medium was removed, 100 µL of Hanks Balanced Salt Solution at room temperature (HBSS; Gibco, Grand Island, NY) was added to the wells and then gently aspirated. Then 100 µL of prepared MTT media (1 mg/mL MTT in culture media) were added to each well. The plates were then incubated for approximately 3 h in a humid ATM containing ~5% CO2 in the air in the dark at 37 ± 2°C. The MTT medium was then removed, and cells were lysed by adding 100 µL isopropanol (Sigma, St. Louis, MO) to each well. Plates were gently shaken for ~20 min on an orbital plate shaker at room temperature. After shaking, absorption was measured at 570 nm with the BioTek Cytation 5 Microplate Reader (Winooski, VT). Cell viability is calculated for each exposure group as a percentage of the mean of the negative control tissues (after subtraction of the background).
Interpretation
Calculation of Cell Viability
Optical density (OD) readings were transferred into Microsoft Excel to allow further calculations to be performed.
The corrected OD (ODc) for each sample or control was calculated by subtracting the value of the blank mean (ODbl) from each reading (ODraw). ODc = ODraw – ODbl.
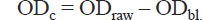
The OD value representing 100% cell viability is the average OD of the negative controls (ODlt_u+MTT).
The percentage of viability for each sample and positive control is calculated as follows:
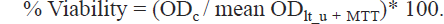
Statistics
For each concentration, the mean, standard deviation, standard error, and percentage of coefficient of variation were calculated in Microsoft Excel. One-way analysis of variance (ANOVA) calculations with Holm–Šídák multiple comparison tests were performed to determine if viability was significantly different in control versus treatment and with different concentrations of H4CBD. ANOVA calculations were performed in GraphPad Prism 9.
AMES Test Method
A plate incorporation assay will be performed using H4CBD (lot# 1.1.22-A5) at concentrations of 1, 5, 10, 50, 100, 500, 1000, and 5000 µg/plate. Plates and assay conditions will be similar to those used routinely during standard bacterial mutagenesis testing. Concentrations may change if solubility issues arise and will be noted in the raw data. If necessary, the dosing solution may be heated to 37°C to help with solubility. The vehicle control will be dimethylsulfoxide (DMSO). The positive control with metabolic activation will be 2-aminoanthracene and without metabolic activation will be 2-nitrofluorene (2NF) (TA98), sodium azide (SA) (TA100 and TA1535), ICR-191 Acridine (ICR) (TA1537), and 4-nitroquinoline-N-oxide (NQNO) (WP2 uvrA). Each concentration of the test article, the vehicle, and the positive controls will be tested in duplicate, both with and without metabolic activation.
Plate Incorporation Procedure
Briefly, either 0.1 mL of the test article or vehicle control, 0.05 mL of positive control, 0.5 mL of S9 mixture (with metabolic activation) or phosphate-buffered saline (without metabolic activation), and 0.1 mL of bacterial culture are added to sterile culture tubes containing 2 mL of supplemented top agar. The mixture is vortexed and poured onto a minimal glucose plate, and the plates are incubated at 37°C for 2 days. After 2 days, the plates are scored for the number of colonies, presence of precipitates (Ps), and confluency of the background lawn.
Test and Control Articles: hERG Patch Clamp
Formulation
Test article solutions will be prepared daily. Test article concentrations will be prepared by diluting stock solutions into an appropriate 4-(2-Hydroxyethyl)piperazine-1-ethane-sulfonic acid (HEPES)-buffered physiological saline solution (HB-PS). Since previous results have shown that ≤0.3% DMSO does not affect channel current, all test and control solutions will contain 0.3% DMSO. The solubility of the test articles at the final concentration of DMSO will be the sponsor’s responsibility. Each test article formulation may be sonicated (Model 2510/5510, Branson Ultrasonics, Danbury, CT) at room temperature to facilitate dissolution. In preparation for the recording session, a glass-lined 96-well compound plate will be loaded with the appropriate amounts of test and control solutions and placed in the plate well of the QPatch HT® or QPatch HTX® (Sophion Bioscience A/S, Denmark).
Positive Control Articles
hCav 1.2 Positive Control
Stock solutions of the positive control article will be prepared in batches, stored frozen, and used within 6 months. The positive control concentration will be prepared fresh daily by diluting stock solutions into HB-PS. The final DMSO concentration will be 0.3%. Name: Nifedipine; source: Sigma-Aldrich; molecular weight: 346.33 g/mol; storage conditions: refrigerated; rationale for selection: previous results have shown that nifedipine at 1 µM blocks hCav1.2 current by approximately 90% (data on file at Charles River Laboratories).
Late hNav 1.5 Positive Control
Stock solutions of the positive control article will be prepared in batches, stored frozen, and used within 6 months. The positive control concentration will be prepared fresh daily by diluting stock solutions into HB-PS. The final DMSO concentration will be 0.3%. Name: Lidocaine; form: HCl salt; source: Sigma-Aldrich; molecular weight: 234.34 g/mol; storage conditions: room temperature; rationale for selection: previous results have shown that 2 mM lidocaine blocks hNav1.5 current by approximately 50%–80% (data on file at Charles River Laboratories).
Cloned Ion Channel Test Systems
Cells will be maintained in tissue culture incubators per Charles River Laboratories SOP. Stocks will be maintained in cryogenic storage. Cells used for electrophysiology will be plated in plastic culture dishes or frozen plugs.
HEK293 Cells
Organism: Homo sapiens, Designation: HEK293, Tissue: Kidney; Transformed with adenovirus 5 DNA; Transfected with ion channel cDNA, Morphology: Epithelial, Age/Stage: Embryo, Source Strain: ATCC, Manassas, VA, Source Substrain: Charles River Laboratories.
HEK293 Culture Procedure
HEK293 cells were stably transfected with the appropriate ion channel cDNA(s). Except for cells that have been stored frozen, stable transfectants will be maintained in the culture medium with the appropriate selection pressure and antibiotics.
CHO Cells
Organism: Cricetulus griseus, Designation: CHO, Tissue: Ovary; transfected with ion channel cDNA, Morphology: Epithelial, Age/Stage: Embryo, Source Strain: ATCC, Manassas, VA, Source Substrain: Charles River Laboratories.
Test Methods For hCav 1.2 and hNav 1.5
Cell Treatments
All experiments will be performed at room temperature. Each cell will act as its own control.
Test Article Treatment Groups
Vehicle will be applied via the QPatch robot pipetting system to naive cells for a 5- to 10-min exposure interval. After vehicle application, the test article concentrations will be applied in at least 3-min intervals (n = 3, where n is the number of cells/concentration). Each solution exchange on the QPatch will be performed multiple times, which will result in 100% replacement of the compound in the QPlate.
Positive Control Treatment Groups
Positive control will be applied in the same manner as the test article to verify sensitivity to ion channel blockade.
Automated Patch Clamp Procedures
In preparation for a recording session, the intracellular solution will be loaded into the intracellular compartments of the QPlate, and the cell suspension will be pipetted into the extracellular compartments. After the establishment of a whole-cell configuration, membrane currents will be recorded using up to 48 parallel patch clamp amplifiers in the QPatch HT® or QPatch HTX® system. Valid whole-cell recordings will meet the following criteria: (1) membrane resistance ≥200 MΩ and (2) leak current ≤25% channel current or subtracted.
Intracellular Solution
Solutions will be prepared in batches, aliquoted, and stored frozen until use. A fresh aliquot will be thawed each day. The pH will be adjusted to 7.2 with N-methyl-
Composition of the Intracellular Solution
hCav1.2: Cs-Asp (130 mM), magnesium chloride hexahydrate (Mg2Cl) (5 mM), ethylene glycol tetraacetic acid (EGTA) (5 mM), Na2-ATP (4 mM), Tris-guanosine 5′-triphosphate (0.1 mM), and HEPES (10 mM).
Nav1.5: cesium fluoride (120 mM), Mg2Cl (2 mM), EGTA (10 mM), cesium chloride (30 mM), sodium fluoride (5 mM), and HEPES (5 mM).
hCav 1.2 Test Procedures
Onset and steady-state block of hCav1.2/β2/α2δ channels will be measured using a stimulus voltage pattern consisting of a depolarizing test pulse (duration, 150 ms; amplitude, 0 mV) at 5 s intervals from a −40 mV holding potential. Peak current will be measured during the step to 0 mV. Leak current will be measured after applying a saturating concentration of cadmium (200 µM) at the end of each experiment to completely block hCav1.2 current.
Late hNav 1.5 Test Procedures
Cells stably expressing hNav1.5 will be held at −80 mV. Onset and steady-state block of late hNav1.5 current due to the test article will be measured using a pulse pattern consisting of a hyperpolarizing conditioning pulse (−120 mV amplitude, 200 ms duration) followed immediately by a depolarizing pulse to −20 mV amplitude, 500 ms duration). This pulse pattern will be repeated at 10-s intervals. At least 100 nM ATXII will be used to elicit late sodium current. Leak current will be measured after applying a saturating concentration of lidocaine (2 mM) at the end of each experiment to completely block late Nav1.5 current.
Data Analysis
A steady state is defined by the limiting constant rate of change with time (linear time dependence). The steady-state before and after the test article application will be used to calculate the percentage of current inhibited at each concentration.
Results and Discussion
Cell Viability Assay Using Primary Lung Fibroblasts (NHLF)
NHLF plays a role in the remodeling of the airway (alveolar structure) as well as airway inflammation; these cells are commonly employed in lung cancer studies due to their prominence in disease progression. Monitoring the NHLF, the number of viable cells in culture was based on the quantitation of ATP present, an indicator of metabolically active cells. H4CBD was compared to a negative control (1% DMSO) and a positive control (1% SDS in media) with a concentration ranging from 1.56 to 50 µM. As shown below in Figure 1a–c, the range of testing for NHLF and other cell lines aside from hERG ranged from 24 to 72 h for detailed results. At the 24-h mark, at the lowest concentration, cell viability was high. With increasing concentrations, cell viability did decrease at the 48- and 72-h marks, as shown below. At lower concentrations, the cell viability remained close to the same as at the 24-h mark. Sustained micromolar concentration over 24 h is the only way to start inducing cytotoxic effects, effectively making low concentrations sustained safe and corresponding to cell viability at given concentrations. Some significant cytotoxicity was observed in NHLF cell lines, starting at 1.56 µM at 24 h and sustained at the 48-h mark. Near total loss of cell viability was observed at elevated concentrations above 1.56 µM, especially with sustained and extended exposure time. The drop in viability from the negative control was significant for all concentrations greater than or equal to 3.13 µM.
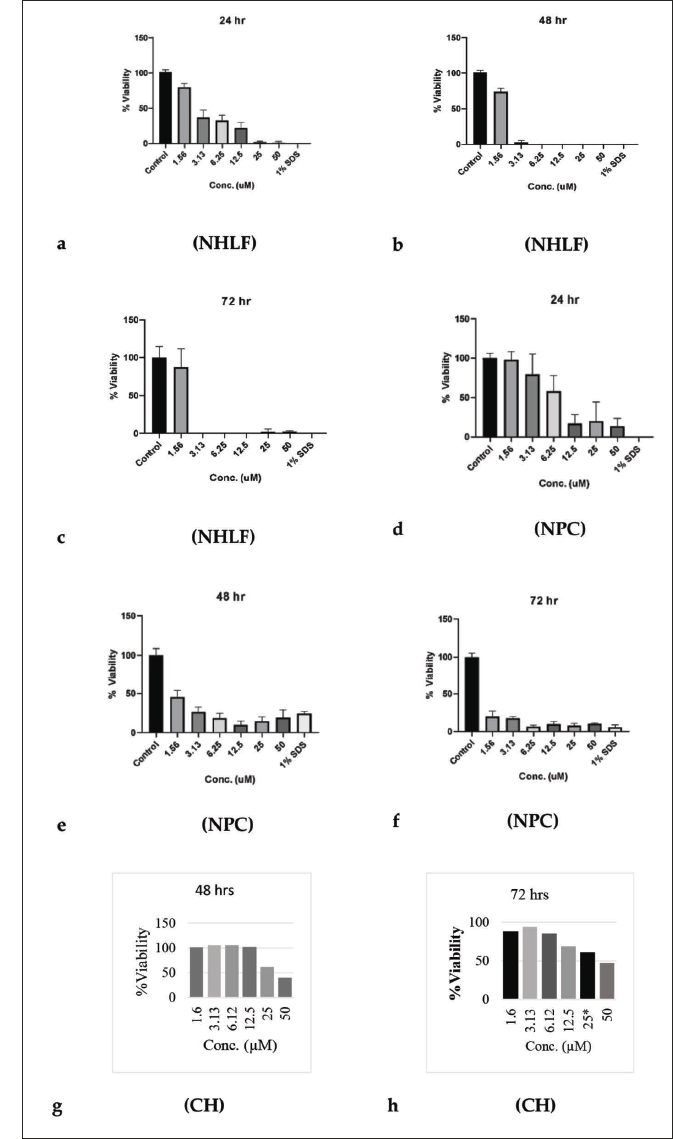
Cell Viability Assay Using Primary Human NPC
Human NPC gives rise to the glial and neuronal cells that populate the central nervous system (CNS), but they do not assist in the generation of nonneural cells. H4CBD did not exhibit low cell viability for NPC at the 24-h mark until the 6.25 µM concentration, as shown in Figure 1d–f below. Cell viability remained low, as depicted at the 48-h mark across all concentrations of H4CBD. Severe loss of cell viability is observed during the 72-h period, with all concentrations exhibiting the same cellular viability characteristics. The observed half-life of cannabinoids falls below the 24-h mark of cell viability limitations. The sustained high concentrations contribute to the loss of cell viability, noting that continuously elevated concentrations would be improbable.
Cell Viability Assay Using Cultured Human Hepatocytes
Hepatocytes play a huge role in a complex system that facilitates the purification of blood components and external contaminants as well as the endocytic uptake of nutrients and needed trophic agents. The in vitro assessment of H4CBD toward the toxicity of cultured hepatocytes was conducted, as shown in Figure 1g and h below; H4CBD did not show any significant drop in cellular viability across varying concentrations at varying times compared to the control. At the 24-h mark across multiple concentrations, cell viability remained constant (data not shown), and drops in cell viability were not noticed across concentrations until the 48- and 72-h marks (Figure 1g and h). Cell viability remained high at 6.25 and 25 µM both at 48 and 72 h, which remains of interest. However, at both 48 and 72 h, there is a trend toward lower viability at 25 and 50 µM concentrations. The speculation for the slight deviation of the hepatocyte at the given time marks is due to slight counting errors when analyzing the well plates for colony reduction shown in Figure 1h.
hERG Assay
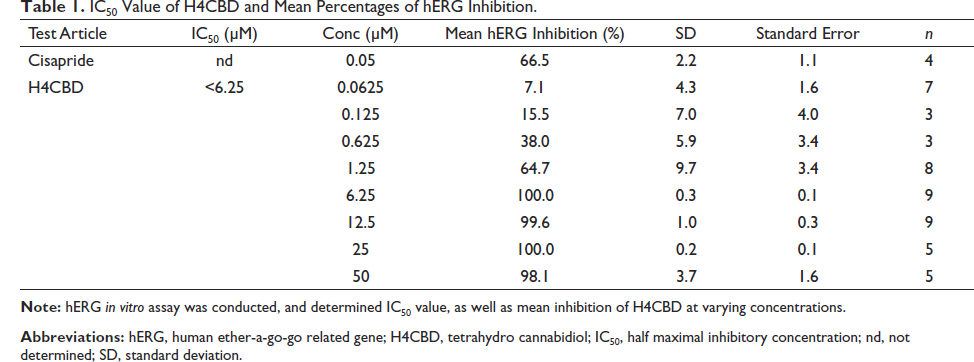
The hERG assay was performed on H4CBD to determine that QTc interval prolongation resulting in potentially fatal ventricular tachyarrhythmia does not occur from any inhibition of ion channels within the heart, including to evaluate anticipated cardiovascular effects from early evaluation of hERG toxicity (Garrido et al., 2020; Meyer et al., 2004; Walker et al., 1999). The hERG study used cisapride as a positive control to examine the effects of H4CBD on cloned hERG potassium channels, encoded by the KCNH2 gene (Oshiroet al., 2010) and expressed in HEK293 cells (Abaandou et al., 2021). H4 CBD was exposed to hERG at 0.0625, 0.125, 0.625, 1.25, 6.25, 12.5, 25, and 50 µM concentrations (n ≥ 3). The duration of exposure to each test concentration was a minimum of 3 min. The positive control data confirmed the sensitivity of the test systems to ion channel inhibition.
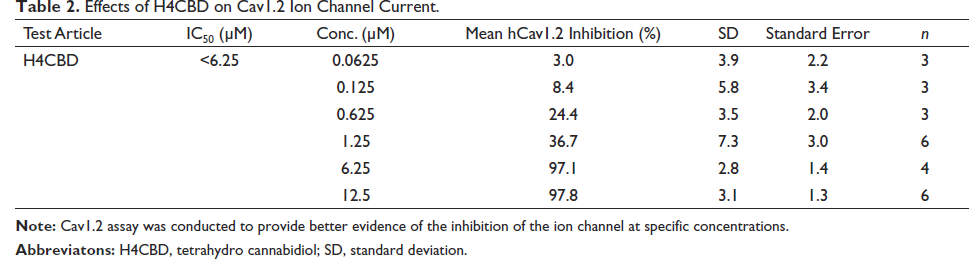
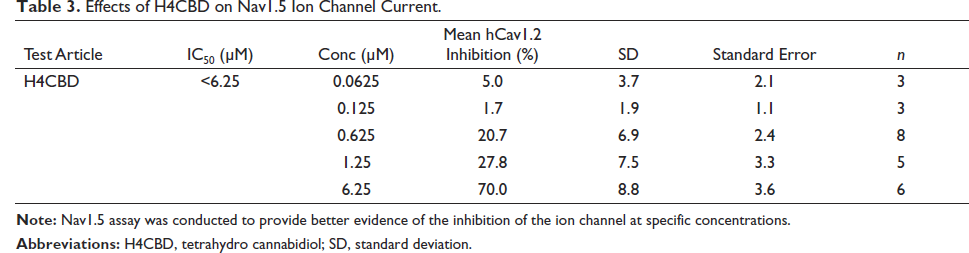
Results shown in Table 1 suggest that H4CBD does not block the hERG-encoding channels that are expressed in the HEK293 cells until above 1.25 µM concentrations. To determine if H4CBD affects other channels, Cav1.2 (Yang et al., 2011) and Nav1.5 (Rook et al., 2012) assays were conducted. The results showed that H4CBD inhibited Cav1.2 calcium and Nav1.5 sodium channels with the same effect as the hERG potassium channels, as shown in Tables 2 and 3. This effect of the depolarization and repolarization by these channels has values that result in a net zero inhibition (Klabunde, 2017; Walker et al., 1999), therefore not inhibiting sodium or potassium ion channels.
Effects of H4CBD on Cav1.2 Ion Channel Current.
Effects of H4CBD on Nav1.5 Ion Channel Current.
AMES Assay
The AMES test (Föllmann et al., 2013) is commonly used, which utilizes bacteria to test whether a compound causes mutations in the DNA of the test organism. Positive results from the test will indicate if the compound is mutagenic or may act as a carcinogen. Thus, the AMES test serves as a quick and convenient assay to estimate the genotoxic potential of a compound.
The data obtained from the AMES study done on the mutagenesis of H4CBD in Escherichia coli is displayed in Table 4. The conducted AMES test resulted in precipitation observed at very high concentrations of drug (≥500 µg/plate) in strains TA98, TA98, TA100, TA1535, and TA1537 with and without S9, and strain WP2uvrA without S9, and at 5000 µg/plate in strain WP2uvrA with S9. Cytotoxicity was observed at ≥5 µg/plate in strains TA100, TA1535, and TA1537 without S9; at ≥50 µg/plate in strains TA100 and TA1537 with S9 and in strain TA98 without S9; at ≥100 µg/plate in strain TA1535 with S9; at ≥500 µg/plate in strain TA98 with S9; and at 5000 µg/plate in strain WP2uvrA without S9. The vehicle and positive controls responded appropriately.
AMES Test on Various Escherichia coli Strains and their Accompanying Dose.
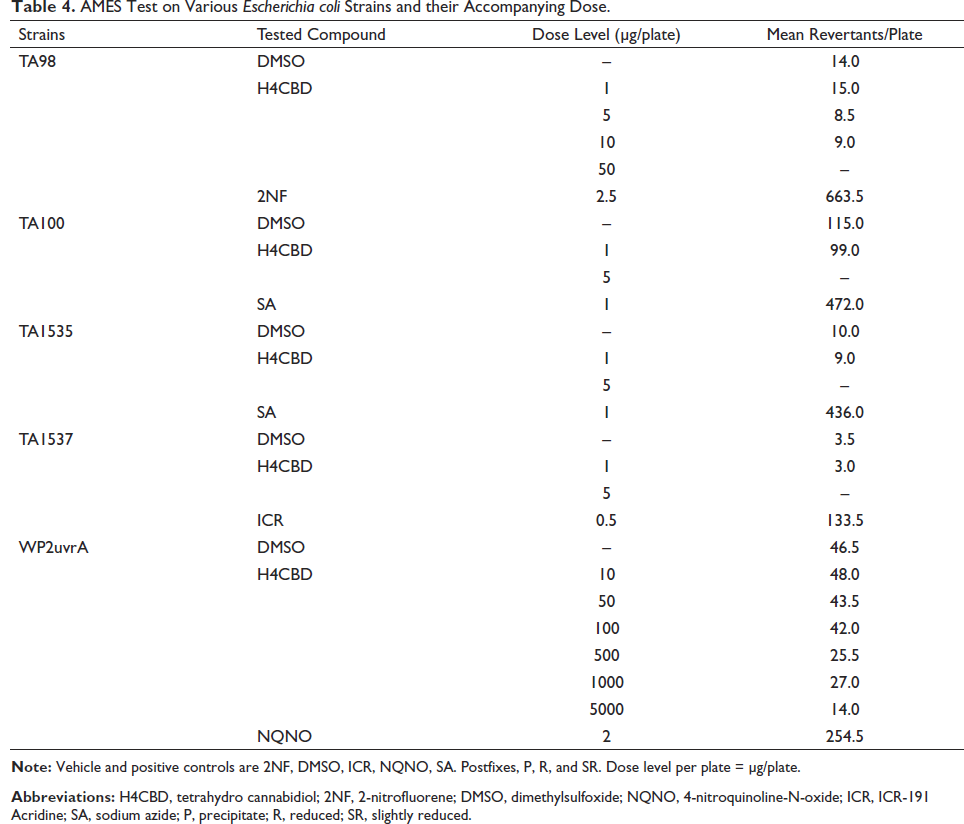
There were no increases in the mean number of revertant colonies as compared to the vehicle control in any strain. Thus, H4CBD is not mutagenic up to cytotoxic concentrations in strains TA98, TA100, TA1535, and TA1537 with and without S9, in strain WP2uvrA with S9, and up to precipitating concentrations in strain WP2uvrA without S9.
Conclusion
The analysis of the different cell types revealed varying responses to H4CBD. NHLF exhibited a concentration-dependent reduction in cell viability, with sustained concentrations over 24 h at 6.25 µM resulting in a significant loss of viability. Conversely, hepatocytes showed a trend of decreased viability at longer exposure times and higher concentrations, but severe cytotoxicity was not observed. This suggests that hepatocytes are less susceptible to the cytotoxic effects of H4CBD compared to NHLF.
In the hERG assay, H4CBD did not inhibit the action potentials within cardiomyocytes, indicating no inhibition of ion channels involved in cardiac function. This finding is important for assessing the potential cardiovascular effects of H4CBD.
The AMES test results were negative, indicating that H4CBD did not demonstrate mutagenic activity in the tested strains. This supports the conclusion that H4CBD does not possess carcinogenic potential based on this assay.
Both human NPC and NHLF showed a median concentration of 3.25 µM before exhibiting a significant reduction in cell viability. This information is valuable for determining research or consumer usage limits for H4CBD.
It is important to note that this study represents a preclinical assessment, and further studies are required. The experimental design followed protocols typically used for preclinical assessments of novel pharmaceuticals. These findings provide insight into the cytotoxic effects of H4CBD and contribute to establishing research and safety parameters as these compounds continue to gain attention.
Abbreviations
HPLC: high-performance liquid chromatography; NMR: nuclear magnetic resonance; MeCN: acetonitrile; DCM: dichloromethane; ATM: atmosphere; CBD: cannabidiol; Pd/C: palladium on carbon; HRMS: high resolution-mass spectrometry; GCMS: gas chromatography-mass spectrometry; 2NF: 2-nitrofluorene; DMSO: dimethylsulfoxide; ICR: ICR-191 acridine; NQNO: 4-nitroquinoline-N-oxide; SA: sodium azide; P: precipitate; R: reduced SR: slightly reduced; NOESY: nuclear overhauser effect spectroscopy.
Footnotes
Acknowledgments
The authors would like to acknowledge Jin Hong of Custom NMR Service for providing professional spectrum analysis and mass spectrometry support from Dr Jasper van Heemst and Scott Caudill at KCA Laboratories in Nicholasville, KY.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors received no financial support for the research, authorship and/or publication of this article.