
Abstract
Background
Juvenile amyotrophic lateral sclerosis (JALS) is a rare and severe form of motor neuron disease characterized by progressive loss of upper and lower motor neurons with an early onset (<25 years).
Purpose
Due to complex etiology and clinical heterogeneity, it is indispensable to unravel molecular mechanisms underlying JALS pathology. The study aimed to identify disease-specific signatures in a 14-years-old sporadic JALS patient.
Methods
Genomic, transcriptomic, and metabolomic analysis of proband and first-degree relatives (FDR).
Results
Exome sequencing identified a novel de novo frameshift variation (c.1465dupG: p.D490Gfs*26) in the fused in sarcoma (FUS) gene in proband. Interestingly, rare and potentially deleterious, disease-modifying variations in DDHD domain containing 1 (DDHD1) and fibrillin 2 (FBN2) were observed. Differentially expressed genes (DGEs) enriched in neuromuscular transmission and inflammatory response were identified by RNA-sequencing. In addition, alterations in purine and pyrimidine, vitamin B6, and sphingolipid metabolism reflect the involvement of inflammatory process in disease pathobiology.
Conclusion
Our findings suggest the involvement of multiple genetic factors coupled with hampered neuromuscular transmission and systemic inflammation in the onset and disease course of JALS.
Keywords
Introduction
Amyotrophic lateral sclerosis (ALS) is an incurable progressive motor neuron disease resulting in skeletal muscle denervation, paralysis, and ultimately death due to respiratory failure. ALS is an adult-onset disorder with a mean age of onset between 51 and 66 years; 1 however, Indian patients have relatively younger onset.2, 3 In a rare subgroup of patients with onset <25 years is considered as Juvenile ALS (JALS). Similar to ALS, JALS pathology also involves the presence of cytoplasmic basophilic inclusions and disorganized intracellular organelles in spinal motor neurons.4, 5 Most studies encompassing JALS are case reports that have employed targeted sequencing of causative genes and identified monogenic variants. Fused in sarcoma (FUS) is the most common pathogenic gene implicated in JALS.6, 7 FUS is a multifunctional RNA binding protein involved in transcription, pre-mRNA splicing, microRNA processing, RNA transport, and maintenance of genomic integrity. Apart from FUS, genetic variations in senataxin (SETX), alsin (ALS2), sigma non-opioid intracellular receptor 1 (SIGMAR1), superoxide dismutase 1 (SOD1), serine palmitoyltransferase long chain base subunit 1 (SPTLC1), SPG11 vesicle trafficking associated, spatacsin (SPG11), ubiquilin 2 (UBQLN2), glucosamine (UDP-N-acetyl)-2-epimerase/N-acetylmannosamine kinase (GNE), TAR DNA binding protein (TARDBP), and DDHD domain containing 1 (DDHD1) account for ~40% of JALS cases. 8
Disease course and progression varies depending upon mutated gene where mutations in FUS are associated with an aggressive disease progression with shorter survival rate (5–24 months)9–11 and mutations in SETX, SPG11, and DDHD1 displayed slower progression rate with prolonged survival (84–276 months).12–14 Given the complex etiology, it is necessary to use multi-pronged strategies to gain insights into pathomechanism of JALS.
In this study, alterations at genetic, transcript, and metabolite levels were analyzed in a 14-years-old sporadic JALS patient and healthy first-degree relatives (FDR). A novel de novo variation in FUS gene was identified in proband by exome sequencing. In addition, rare and potentially deleterious, disease-modifying mutations were inherited in proband. Transcriptome analysis highlighted the role of synaptic transmission, axonal growth and guidance, muscle contraction, and systemic inflammation in JALS. Further, altered metabolic signatures suggested involvement of purine and pyrimidine, vitamin B6, and sphingolipid metabolism in disease mechanisms.
Methods
Subjects
A 14-year-old sporadic juvenile ALS patient (proband), healthy FDR (father, mother, and sister) and an unrelated age/gender matched healthy control (referred as control) were enrolled in the study. Procedure for isolating DNA, RNA, and serum are provided in Supplementary methodology. Study was approved by the Institutional Ethics Committee (EC/05/20/1714). Written informed consent was obtained from all participants. Study was conducted according to the principles of the Helsinki Declaration of 1964, as revised in 2013.
Whole Exome Sequencing
Whole exome sequencing was performed using the Twist Comprehensive Exome Panel. DNA libraries were indexed, pooled, and sequenced on Illumina NextSeq 550 Platform. To analyze data, three gene sets were defined: (1) genes associated with JALS/ALS; (2) genes associated with ALS-mimicking disorders, neurodegenerative disorders, and/or other related neuromuscular disorders; and (3) Rare Variants. Intolerance to functional variants for the identified genes was also estimated by Residual Variation Intolerance Score (RVIS), probability of being loss of function intolerant (pLI), and missense z conservation scores (
RNA Sequencing and Pathway Analysis
The mRNA sequencing was performed using Illumina HiSeq 4000 system (2 × 150 paired end). Nextflow RNA-Seq (nf-core) pipeline was used for analysis. Differentially Expressed Genes (DEGs) and visualizations were done using the iGEAK RNA-seq software (Supplementary Figure 1, Supplementary Tables 1 and 2). Pathway enrichment of DEGs was performed using Ingenuity Pathway Analysis (IPA) and Enrichr. Interaction of gene-pathway networks was visualized using ClueGO Cytoscape plugin. Details are provided in the Supplementary methodology. Genes identified in RNA-seq analysis were validated by real-time quantitative reverse transcription PCR (qRT-PCR) using SYBR green.
Metabolomics and Pathway Enrichment
Matyash method was used for metabolite extraction with minor modifications. Extracted metabolites were separated in Acquity UPLC HSS T3, a reverse-phase column connected with Dionex Ultimate 3000, liquid chromatography. Details for acquisition, pre-processing, and identification of differentially regulated metabolites are provided in the Supplementary methodology. Metabolic pathway analysis was carried out using MetaboAnalyst 5.0.
Results
Clinical Findings
A 14-year-old boy was first seen at neurology outpatient service on 5 June 2021 with onset of symptoms including weakness, wasting, and fasciculation of distal muscles of the right upper limb at the age of 13 years. Disease rapidly progressed with weakness in proximal muscles of right upper limb, weakness of lower limbs, trunk and neck muscles followed by head drop, slurred speech, and difficulty in swallowing food. He was fully dependent for all activities of daily living and was in a wheelchair.
There is no history of motor neuron disease, Parkinson’s disease, or Alzheimer’s disease in the family for three generations and the sibling is normal (Figure 1A). He was a product of non-consanguineous marriage and normal delivery. Motor mile stones and speech were delayed along with learning disability. Neurological examination revealed weakness of gag reflex, normal tongue movement with no evidence of atrophy or fasciculations. There was atrophy, weakness, fasciculations and minipolymyoclonus of both hands with changes being more marked on the right side. Distal muscles were more affected than proximal muscles in both upper and lower limbs. Jaw jerk was absent, palmomental reflex was present, and deep tendon reflexes in upper limbs were absent while they were exaggerated in lower limbs with extensor plantar response.
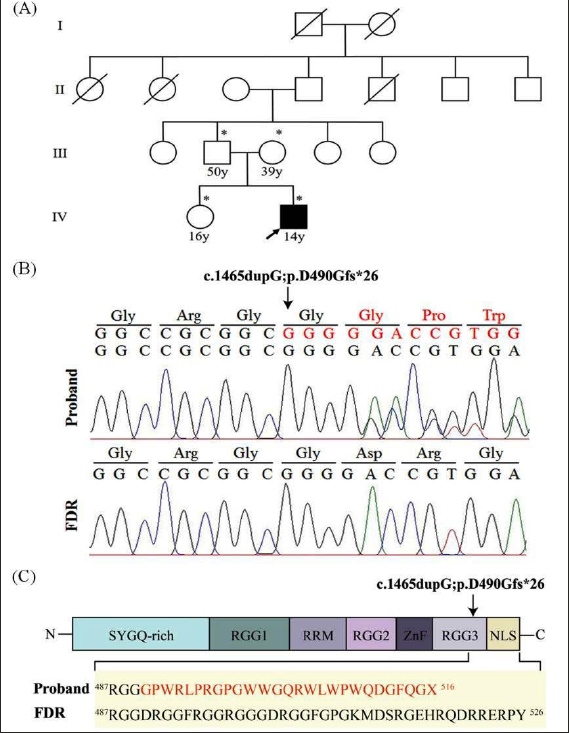
De novo Variation in FUS Identified in Juvenile ALS. (A) Pedigree Chart of Sporadic Juvenile ALS Patient (□: Males; ○: Females; ■ Affected Individual; Diagonal Lines Across Symbols Indicate Deceased Individuals; Arrow Indicates the Proband). The Family Quartet Analyzed in the Study is Marked with *. (B) Validation of Wild Type and Novel FUS Mutation by Sangers Sequencing in Proband and FDR (First degree Relative). Respectively. (C) Schematic Representation of the Structure of FUS Protein, Highlighting Novel De-novo Mutation Identified in Proband. Arrow Shows Position of Mutation in (B) and (C) Leading to Altered DNA/Amino Acid Sequence (represented in red) in Proband.
All routine blood tests were normal. Nerve conduction studies showed distal asymmetrical large fiber motor axonal polyneuropathy involving both upper and lower limbs, changes more marked on right compared to left limbs, and no conduction block. Electromyography showed acute and chronic denervation in distal and proximal muscles of all four limbs and cervical paraspinal muscles. Thoracic paraspinal and cranio-bulbar muscles did not show any abnormality. Findings were consistent with preganglionic involvement suggestive of anterior horn cell disorder. Based on revised El Escorial criteria, diagnosis of definite amyotrophic lateral sclerosis was considered. Supplements, riluzole, and supportive treatment were given. There was progressive deterioration with breathlessness in December 2021 necessitating tracheostomy with ventilator support. The ALS-functional rating scale revised (ALSFRS-R) score was 23. The period from onset of symptoms to tracheostomy was 20 months.
Deleterious Genetic Variations Associated with Juvenile ALS
In order to identify genetic factors associated with disease etiology, exome sequencing of family quartet was carried out and a novel de-novo variation (c.1465dupG: p.D490Gfs*26) in exon 14 of FUS gene was identified in the proband (variant submitted to ClinVar, accession number SCV003842176). The pathogenic mutation in FUS translates (in silico) into a truncated protein with altered C-terminal nuclear localization signal (NLS) (Figure 1B). Another novel variation of uncertain significance (c.A2110C: p.N704H) in exon 10 of DDHD1, the gene previously implicated in JALS was identified in proband. 14 Further, rare deleterious variations in fibrillin 2 (FBN2), hexokinase domain containing 1 (HKDC1), fibrillin 1 (FBN1), fibrinogen C domain-containing protein 1 (FIBCD1), SEC23 homolog B (SEC23B), and serpin A12 (SERPINA12) genes were also observed in proband. Moreover, according to constraint metrics, FUS, DDHD1, and FBN2 genes qualified as intolerant to missense variations (Table 1).
Deleterious Variants Identified in Juvenile ALS.
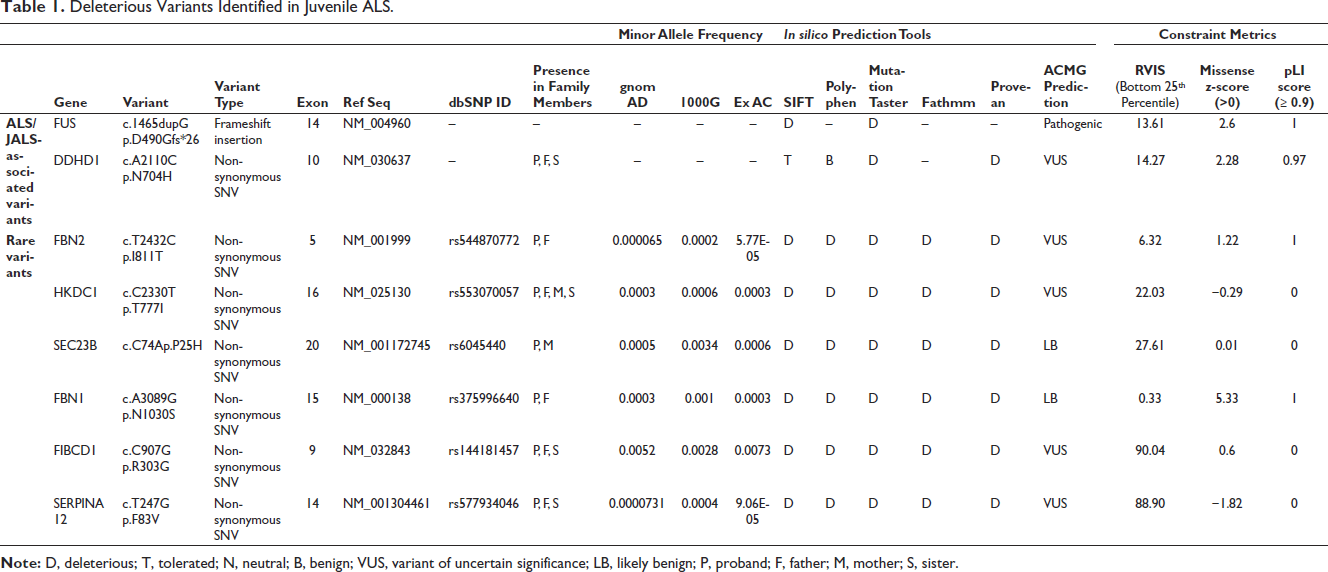
Apart from FUS, at least one of the family members was positive for other predicted pathogenic variations identified in proband (Table 1). Another variant of interest, c.2330C > T in HKDC1, was homozygous in proband and heterozygous in family members. Pathogenic repeat expansion in C9orf72 was absent in proband (data not shown) as previously observed in ALS patients from India. 15 These deleterious variations were absent in 40 healthy controls. Further, no pathogenic variation was observed in any other ALS and neuromuscular disorder-associated genes. Thus, FUS appears to be the causative gene and other inherited variations may act as disease modifiers.
Altered Transcriptome Profile Enriched in Inflammation and Neuromuscular Synaptic Transmission
In order to examine the impact of FUS and other deleterious variations, transcriptome analysis of the family quartet was carried out from the peripheral blood mononuclear cells (submitted to GEO repository, accession number GSE232929).
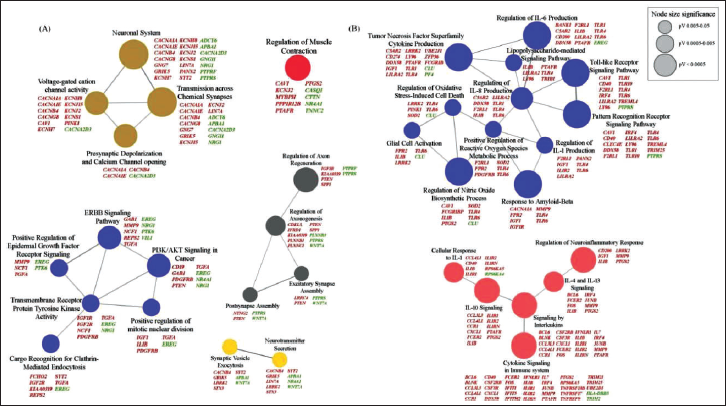
IPA of all DEGs (Figure 2A and B, Supplementary Tables 3 and 4) showed activation of cAMP response element-binding protein (CREB) signaling in neurons suggesting abnormal neuronal excitation, metabolism, synaptic plasticity, and survival. Pathway enrichment using Enrichr of 784 mutual (P vs. FDR and P vs. control) DEGs (Figure 2A and C, Supplementary Table 5) and network construction using ClueGO revealed several pathways involved in neuromuscular transmission (Figure 3). Alterations in calcium and potassium voltage-gated ion channel genes reflected abnormal neuronal excitability. Levels of sodium channel genes (sodium voltage-gated channel beta subunit 1 (SCN1B) and sodium voltage-gated channel alpha subunit 3 (SCN3A)) were also observed to be altered. Genes (amyloid beta precursor protein binding family A member 1 (APBA1), LIN7A, leucine-rich repeat kinase 2 (LRRK2), syntaxin 3 (STX3), and synaptotagmin 2 (SYT2)) involved in docking, fusion, and release of neurotransmitter displayed deregulated transcript levels indicating abnormal release of neurotransmitter. Pathways related to axon growth and guidance were also enriched. Downregulation of neuregulin 1 (NRG1) and Ly6/neurotoxin 1 (LYNX1) may affect the functional activity of nicotinic acetylcholine receptors (nAChRs). Further, regulation of skeletal muscle contraction was also affected due to altered levels of pivotal genes including dysferlin (DYSF), myosin binding protein H (MYBPH), calsequestrin (CASQ1), and troponin C2 (TNNC2). Neuromuscular transmission genes were validated using qRT-PCR (Supplementary Figure 2).
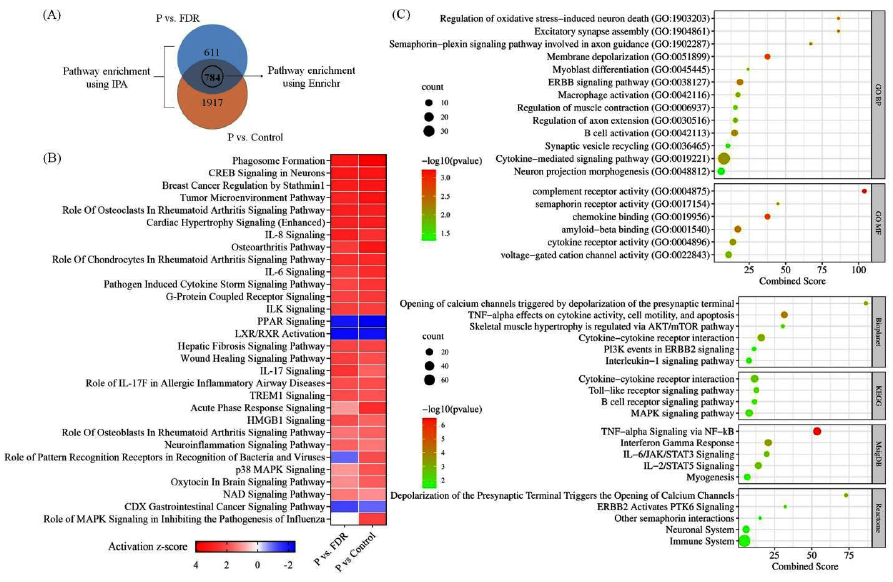
In addition, activation of interleukin (IL-6, IL-8), cytokine, triggering receptor expressed on myeloid cells 1 (TREM1), and neuroinflammation signaling pathways reflected a major involvement of systemic inflammation in disease pathology. Further, downregulation of peroxisome proliferator-activated receptor (PPAR), liver X receptor/ retinoid X receptor (LXR/RXR), interleukin-4 (IL-4), interleukin-10 (IL-10), and interleukin-13 (IL-13) signaling pathways indicated suppression of anti-inflammatory response (Figures 2B, C and 3).
Altered Metabolic Signature in Juvenile ALS
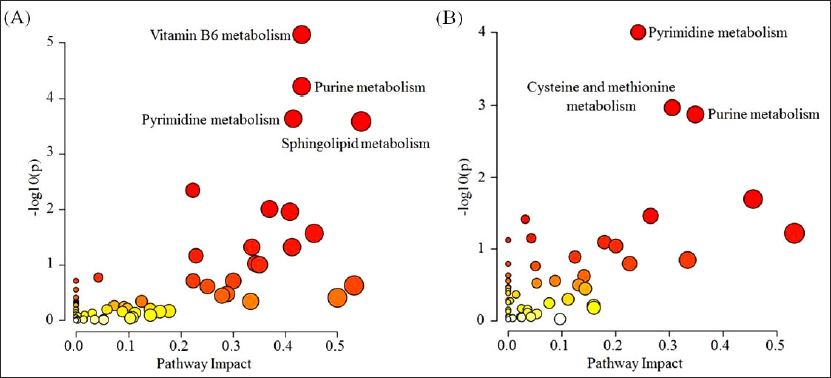
With the basic assumption of relative expression ordering analysis (REOA), where a pair of features holds same order relationship (a < b or a > b) under normal physiology, whereas this order alters in pathological states, highly stable metabolic feature pairs with consistent REO among FDR and control reported 2,77,320 and 9,49,130 stable pairs identified in both RP positive and negative modes, respectively. Using these reference stable pairs, dysregulated metabolic feature pairs were assessed for all case control situations as described in methodology. A total of 4398 metabolic features (1552 features from RP positive; 2846 features from RP negative) were dysregulated in the comparison groups. These significant features were mapped to 1592 compounds following metabolite annotation and identification of metabolites related to endogenous human metabolic pathways. Metabolic pathway analysis of all annotated compounds dysregulated in proband compared to FDR and control mapped to 62 kyoto encyclopedia of genes and genomes (KEGG) metabolic pathways (Supplementary Tables 6 and 7). Among those, four pathways namely vitamin B6 metabolism, purine metabolism, pyrimidine metabolism, and sphingolipid metabolism demonstrated significant changes in proband compared to FDR (Figure 4A, Table 2). While comparison between proband and control showed significant changes in purine, pyrimidine, and cysteine and methionine metabolic pathways (Figure 4B, Table 2).
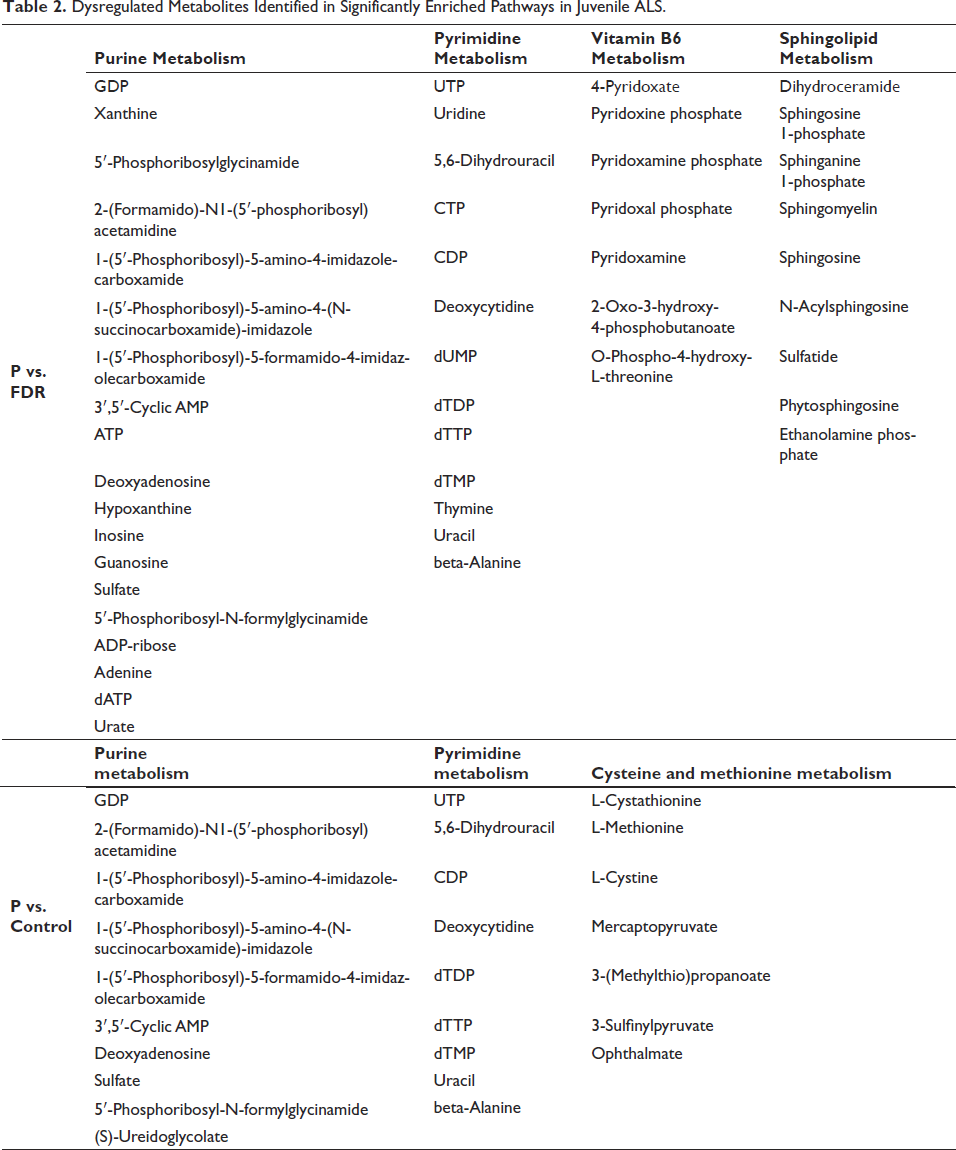
Dysregulated Metabolites Identified in Significantly Enriched Pathways in Juvenile ALS.
Discussion
In the past decade, advancement in omics technology has significantly enhanced understanding of complex etiology of ALS. However, no multiomics study has been carried out to identify disease signatures in JALS. This study has investigated genetic, transcriptomic, and metabolomic alterations in peripheral blood of a JALS patient. A novel de novo mutation in arginine–glycine–glycine repeat domain (RGG3) domain of FUS resulting in altered NLS and premature truncation was identified in proband. Most of the known FUS variations are mapped in or near C-terminus affecting NLS leading to cytoplasmic mislocalization. Mutant FUS accounts for >90% of sporadic JALS cases 7 and <1% of sporadic adult-onset ALS. 16 FUS-associated JALS patients displayed median age of onset of 18 years with an average disease duration of 12 months, 7 whereas adult-onset patients showed median age of onset of 39 years with an average disease duration of 25 months. 17 Different mutations in FUS displayed distinct clinical disease parameters including age/site of onset and onset to severe event (ventilation support, tube feeding, or death). 17 Such clinical heterogeneity may involve influence of other genetic factors. Apart from FUS, proband inherited rare damaging variations in DDHD1 and FBN2 genes, which may act as disease modifiers. DDHD1 is a phospholipase, implicated in lipid metabolism and mitochondrial functioning, and hence, it may have a role in ALS pathogenesis. 18 Previously, missense variation in DDHD1 was identified in a sporadic JALS patient from China. 14 FBN2 encodes for fibrillin 2, which self-polymerizes to form microfibrils and involved in extracellular matrix formation and remodeling. 19 A recent study identified variations in genes related to intellectual disabilities in two unrelated JALS patients harboring FUSP525L mutation. 20 In our study, proband displayed learning disabilities but no genetic variations were identified in genes previously implicated in intellectual disability. Moreover, presence of rare variant burden has been shown to influence age of onset and survival in ALS patients.21, 22
It is important to note that ongoing degeneration in primary tissues involved in disease pathology are mirrored in peripheral blood cells, which strengthen its role as a surrogate marker in ALS.23, 24 Malfunctioning of neuromuscular junction (NMJ) is considered as one of the primary events in ALS pathology. 25 FUS NLS mutations have been shown to disrupt axonal transport and cause NMJ defects in primary cultured neurons, 26 transgenic mice and co-culture of induced pluripotent stem cells (iPSCs)-derived motor neurons and myotubes from FUS ALS patients. 27 Transgenic ALS mice expressing mutant FUS displayed altered expression of ion channels and transporters required for synaptic function. 28 Our findings also revealed abnormal expression of genes encoding for voltage-gated ion channels suggesting deranged neuronal excitability in JALS. Further, the presence of altered transcript levels of genes involved in neurotransmitter release and muscle contraction in our study suggests aberrations in neuromuscular transmission, which support results from previous reports on ALS patients29–31 and mutant FUS models.32, 33 Altered levels of NRG1 and LYNX1 observed in proband reflected aberrant synaptic fidelity at the NMJ. NRG1-mediated ERBB signaling has been shown to be altered in ALS 34 and overexpression of NRG1 in muscle of SOD1G93A mice promotes axonal reinnervation and prevents denervation of muscles. 35 Further, changes in level of genes involved in regulation of axon regeneration indicate NMJ remodeling, a compensatory mechanism implicated in disease pathology. 36
Neuroinflammation, characterized by activation of microglia, infiltration of lymphocytes, macrophages, has been well documented in ALS. Transcriptome analysis of spinal cord tissue from post-mortem ALS patients showed over-representation of microglia and upregulation of pathways associated with neuroinflammation and immune response.29, 37 Further, disruption of blood-central nervous system (CNS) barriers in ALS opens the gate for crosstalk between resident cells of central and peripheral immune system leading to systemic inflammation. 38 In alignment with the previous ALS reports,39–41 RNA-seq analysis in our study showed activation of inflammation and immune response in peripheral blood of JALS patient. Immunomodulatory therapeutic strategies have shown potential in attenuating inflammatory pathology and reducing disease progression in ALS.30, 42
Metabolic profiles of ALS patients vary depending upon rate of disease progression.43, 44 We observed dysregulated metabolic signatures, which highlight systemic inflammation in JALS. Consistent with previous findings, defects in nucleic acid metabolism appear to be the top perturbed pathways in JALS patient. Altered purine metabolism has recently been reported in two JALS patients harboring pathogenic ALS2 mutation. 45 Fibroblast of sporadic ALS patients also showed altered purine and pyrimidine metabolism, which strengths their role in oxidative stress and inflammatory response in ALS. 46 Even expression of SOD1G93A has been shown to alter astrocytes metabolome by dysregulating levels of metabolites involved in purine metabolism. 47 Studies have reported significant lower levels of uric acid, a natural antioxidant and end metabolic product of purine metabolism, in ALS patients compared to healthy controls. This reflects increased oxidative stress as a disease mechanism and there exists an inverse correlation between uric acid levels and disease duration.48, 49
Increased oxidative stress may also induce alterations in sphingolipid metabolism leading to motor neuron degeneration. Impaired sphingolipid metabolism has been observed in proband, which is consistent with previous reports.50–52 Further, dysregulation of Vitamin B6 metabolism was also observed in JALS patient. Vitamin B6, a water-soluble vitamin critically involved in CNS and peripheral nervous system (PNS) functions, has been proposed as a protector of ALS progression. Vitamin B6 lowers elevated homocysteine levels observed in ALS patients 53 by converting it to sulfur amino acids. Moreover, Vitamin B6 supplementation has been shown to reduce inflammation via crosstalk with sphingolipid metabolism. 54 Hence, further studies are warranted to evaluate Vitamin B6 as a palliative therapy for ALS/JALS.
Conclusion
Juvenile ALS is an extremely rare form of ALS, and therefore, access to a larger patient population is not always feasible. Though, sample size is the limitation of the study but multiomics analysis of the family quartet is a step toward a better understanding of the complex molecular etiology of sporadic JALS. Taken together, our data pinpoints the role of rare variant burden, altered neuromuscular transmission, and inflammation in disease pathology. These foundational results need to be validated in a larger patient population, which would open the perspective of novel therapeutic interventions.
Abbreviations
JALS: Juvenile amyotrophic lateral sclerosis
FUS: Fused in sarcoma
SETX: Senataxin
ALS2: Alsin
SIGMAR1: Sigma non-opioid intracellular receptor 1
SOD1: Superoxide Dismutase 1
SPTLC1: Serine Palmitoyltransferase Long Chain Base Subunit 1
SPG11: SPG11 vesicle trafficking associated, spatacsin
UBQLN2: Ubiquilin 2
GNE: Glucosamine (UDP-N-Acetyl)-2-Epimerase/N-Acetylmannosamine Kinase
TARDBP: TAR DNA binding protein
DDHD1: DDHD domain containing 1
FDR: First-Degree Relatives
RVIS: Residual Variation Intolerance Score
pLI: probability of being loss of function intolerant
DEGs: Differentially expressed genes
IPA: Ingenuity Pathway Analysis (QIAGEN IPA)
ALSFRS-R: ALS-Functional Rating Scale Revised
NLS: Nuclear Localization Signal
FBN2: Fibrillin 2
HKDC1: Hexokinase domain containing 1
FBN1: Fibrillin 1
FIBCD1: Fibrinogen C domain-containing protein 1
SEC23B: SEC23 homolog B
SERPINA12: Serpin A12
CREB: cAMP Response Element-Binding Protein (CREB)
SCN1B: Sodium Voltage-Gated Channel Beta Subunit 1
SCN3A: Sodium Voltage-Gated Channel Alpha Subunit 3
APBA1: Amyloid Beta Precursor Protein Binding Family A Member 1
LRRK2: Leucine-rich repeat kinase 2
STX3: Syntaxin 3
SYT2: Synaptotagmin 2
NRG1: Neuregulin 1
LYNX1: Ly6/neurotoxin 1
nAChRs: Nicotinic Acetylcholine Receptors
DYSF: Dysferlin
MYBPH: Myosin binding protein H
CASQ1: Calsequestrin
TNNC2: Troponin C2
qRT-PCR: Real-Time Quantitative Reverse Transcription PCR
IL-6: Interleukin-6
IL-8: Interleukin-8
TREM1: Triggering Receptor Expressed On Myeloid Cells 1
PPAR: Peroxisome proliferator-activated receptor
LXR/RXR: Liver X Receptor/ Retinoid X Receptor
IL-4: Interleukin-4
IL-10: Interleukin-10
IL-13: Interleukin-13
GO: Gene Ontology
KEGG: Kyoto Encyclopedia of Genes and Genomes
REOA: Relative Expression Ordering Analysis
GDP: Guanosine diphosphate
UTP: Uridine triphosphate
CTP: Cytidine triphosphate
CDP: Cytidine diphosphate
dUMP: Deoxyuridine monophosphate
dTDP: Deoxythymidine 5’-diphosphate
dTTP: Deoxythymidine triphosphate
dTMP: Deoxythymidine monophosphate
RGG3: Arginine–Glycine–Glycine repeat Domain
NMJ: Neuromuscular Junction
iPSCs: Induced Pluripotent Stem Cells
CNS: Central nervous system
PNS: Peripheral nervous system
Footnotes
Acknowledgements
Authors deeply appreciate Kavita Vats and Abhishek Vats for assisting with RNA-seq data analysis. Authors also acknowledge Dr. Renu Saxena for help in C9orf72 repeat expansion.
Authors’ Contribution
SV, SK: conceptualization of the study, genomics, transcriptomics and metabolomics analysis, manuscript writing; YV: validation by Sanger sequencing; AS, MJ and PK: Mass spectrometry and metabolomics analysis; NKG and SW: critical reviewing; PC: conceptualization and design of the study, critical reviewing; IA and MG: clinical examination and diagnosis, manuscript writing and editing; AS: neurophysiological examination; VT: conceptualization and design of the study, omics analysis, manuscript writing, and editing. All authors read and approved the final manuscript.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
This study was supported by the Indian Council of Medical Research (5/4-5/Neuro/215/2020-NCD-1). SV was supported by the Council of Scientific and Industrial Research for SRF-NET scholarship (09/591(0150)/2018-EMR-I). SK was supported by SRF scholarship from the Indian Council of Medical Research (3/1/2/151/Neuro/2021-NCD-I).
ICMJE Statement
In compliance with the ICMJE uniform disclosure form, all authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Statement of Ethics
Study was approved by the Institutional Ethics Committee, Sir Ganga Ram Hospital, Delhi (EC/05/20/1714). Written informed consent was obtained from all participants. Study was conducted according to the principles of the Helsinki Declaration of 1964, as revised in 2013.
Supplemental Material
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.