
Abstract
Ewing’s Sarcomas (ES)/Peripheral neuroectodermal tumour (pPNET) are heterogenous group of rare, highly malignant, undifferentiated primitive round-cell neoplasms of neuroectodermal origin. pPNETs are seldom observed to involve the spine of which Spinal Intradural Extramedullary Extraosseous Primary ES/pPNET are extremely rare. We report a case of a 23-year-old male with complaints of low backache and hip pain radiating to the left inguinal region for four months. Radiology findings were suggestive of a neurogenic tumour. Cytomorphology, histomorphology and immunohistochemistry evaluation were done. Diagnosis was consistent with ES/pPNET. Careful correlation between clinical history, cytomorphology, histopathology, immunohistochemical and molecular analysis can help to distinguish primary spinal ES/PNET from other primary spinal tumours and will help clinicians to start treatment at the earliest.
Introduction
Ewing’s Sarcomas (ES) are rare, highly malignant, primitive, undifferentiated round cell neoplasms of neuroectodermal origin.1, 2 They belong to Ewing’s Sarcoma Family Tumours (ESFTs) and are seen in children and adults. Askins tumours, pPNET, Osseous Ewing’s Sarcoma (OES) and Extraosseous Ewing’s Sarcoma (EES) are included in this spectrum. 3 pPNETs are seldom observed to involve the spine. ES arising from the spine is classified into sacral and non-sacral types. Sacral-type ES is more aggressive and shows poor responses to treatment. The non-sacral ES spine represents approximately 0.9% of all cases. 4 Thus, primary spinal Intradural Extramedullary Extraosseous ES (IEEES) is extremely rare.4–12 We, here, report an unusual, yet the interesting presentation of Primary Intradural Extramedullary ES/pPNET with extradural extension in the lumbar spine.
Case Presentation
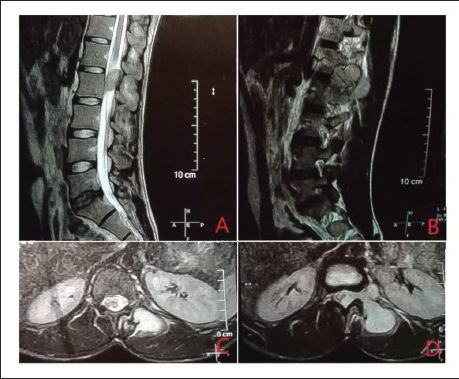
A 23-year-old man presented with complaints of low backache and hip pain radiating to the left inguinal region for four months. The pain was not radiating to the lower limb. Bowel and bladder control was intact. MRI LS spine demonstrated a lobulated T2/STIR hyperintense soft tissue lesion in an intradural location with extension into the extradural space and via L1−L2 and L2–L3 neural foramina into the left paravertebral and paraspinal space (Figure 1). Lesion measures 6.0 × 5.0 × 3.9 cm. Findings were suggestive of an intradural extramedullary neoplastic lesion with a dumbbell appearance suggestive of a neurogenic tumour. MRI Brain was normal ruling out a primary brain lesion. Biochemical tests and CBC were normal.
(A) Sagittal T2-Weighted MRI Image of the Lumbar Spine Reveals a Relatively Well-defined T2 Iso- to Hyperintense Lesion, which is Predominantly Intradural and Extramedullary in Location with Some Regions of Extradural Extension. (B) Sagittal T2-Weighted MRI Image of the Lumbar Spine Shows T2 Iso- to Hyperintense Soft Tissue Extending Out Through the Neural Foramen. (C) T2 Axial Fat Saturated Images Showing a Well-defined T2 Hyperintense Soft Tissue Lesion in the Left Paravertebral Muscles. (D) T2 Axial Fat Saturated Images of the Spine at L2–L3 Level Shows a Dumbbell-shaped T2 Hyperintense Lesion in a Predominantly Extramedullary Intradural Location, with Epidural Extension and Extending Through the Neural Foramina on Left Side into the Left Paravertebral Space.
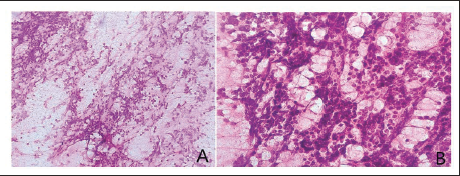
Following hemilaminectomy and Gross Tumour Reduction (GTR), Squash samples were taken and then sent for intraoperative cytological diagnosis. Sample was easy to smear and smears were cellular showing a uniform population of round cells with opened chromatin and scant cytoplasm at places forming small clusters and in a rosette pattern. Few clusters of these cells were entangled in fibrous tissue along with a sprinkling of lymphocytes (Figure 2). An opinion of a small round cell tumour was made on cytology.
Squash Cytology. (A) Population of Round Cells with Entangled in Fibrous Tissue (Giemsa Stain, Original Magnification × 4). (B) Undifferentiated Round Cells with Opened Chromatin and Scant Cytoplasm. Few Clusters Showed Sprinkling of Lymphocytes Tissue (Giemsa Stain, Original Magnification × 40).
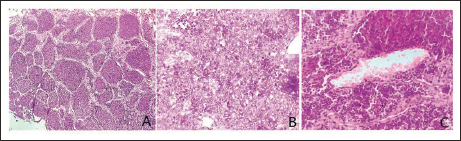
Histopathological examination showed sheets and lobules of undifferentiated small round cells with hyperchromatic round nuclei, scant cytoplasm and separated by thin fibrovascular septae. These tumour cells were seen around blood vessels (Peritheliomatous pattern). Frequent mitosis is seen (Figure 3).
Histopathology. (A) Undifferentiated Small Round Cells Arranged in Sheets and Lobules Separated by Thin Fibrovascular Septae. (B) Undifferentiated Small Cells Having Hyperchromatic Round Nuclei and Scant Cytoplasm with Presence of Frequent Mitosis. (C) Small Round Tumour Cells Showing Peritheliomatous Pattern (H&E Stain, Original Magnification 40 × and × 400 ×).
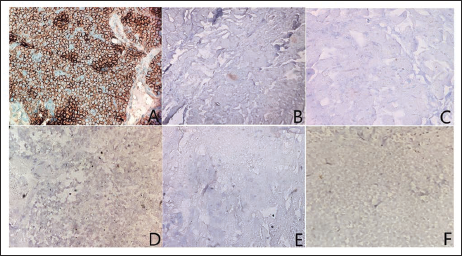
The tumour cells were strongly immunoreactive for CD99 and FLI-1 and patchy positive for vimentin. They were negative for WT1, desmin, p63, LCA, S-100, synaptophysin, CK and vimentin (Figure 4). No cytogenetic or molecular testing was performed. The final histopathology report was a small round blue cell tumour with features compatible with ES/pPNET.
Immunohistochemistry. (A) CD 99 (++), (B) Synaptophysin (–), (C) P63 (–), (D) Desmin (–), (E) WT1 (–) and (F) LCA (–).
Post-operative MRI shows diffusely enhancing lesion measuring 3.0 × 2.0 × 1.5 cm. The patient was given chemotherapy and local radiotherapy and serial MRI were taken at appropriate intervals. The patient clinically improved and was responding well to the treatment and interval reduction was noted. No residual tumour was found after 11 cycles of chemotherapy and radiation treatment (45 Gy in 25 fractions). However, after six months, patient developed disseminated disease with multiple bone and lung metastasis. He was put on metronomic oral chemotherapy and supportive care. Later, patient also developed salivary gland swelling, which on histopathology was suggestive of metastatic deposits of ES.
Implications
Unusual location of ES can hinder the approach to the clinical diagnosis. Careful correlation between clinical history, cytomorphology, histopathology, immunohistochemical and molecular analysis should be done to distinguish primary spinal ES/PNET from other primary spinal tumours.This will help clinicians to start treatment at the earliest.
Discussion
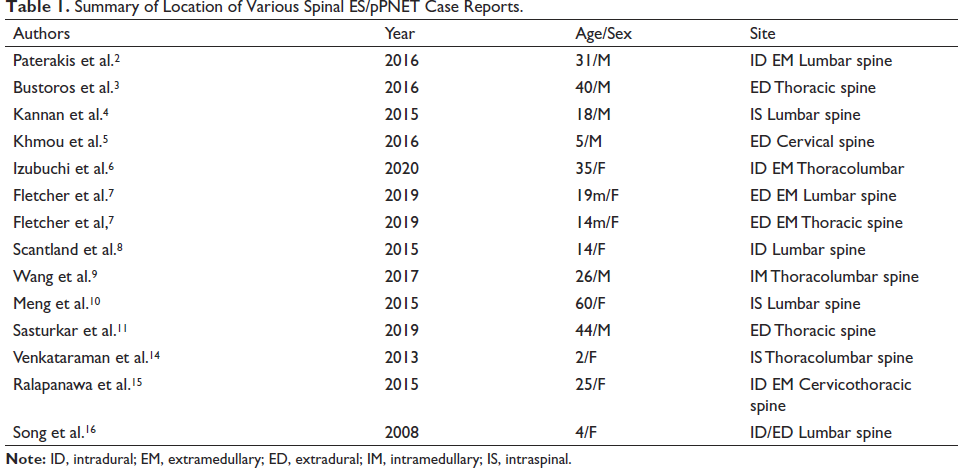
Even though “primitive neuroectodermal tumour” terminology was removed in WHO 2016 classification; this term has been widely used in synonymous with ES in literature.9, 13 ES are classified as small round cell tumours with specific molecular findings and varying neuroectodermal differentiation in immunohistochemistry. 6 Locations such as the extremities, paravertebral and pelvic locations are commonly reported but intradural spinal ES is extremely rare.4, 6, 8–11, 14 In spine, the common site is the lumbar region, followed by thoracic and cervical spine. Sacral region is the least common among spinal ES 3 (Table 1).
Summary of Location of Various Spinal ES/pPNET Case Reports.
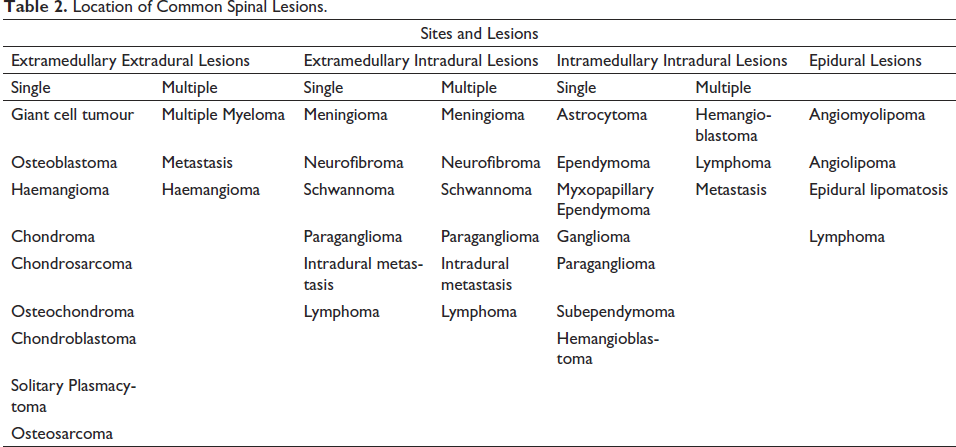
Common Intradural spinal tumours include meningioma, astrocytoma, ependymoma, nerve sheath tumours such as schwannoma or neurofibroma and metastasis (Table 2). The imaging and clinical features of IEES in the initial stage mimic abovementioned intradural spinal tumours. Therefore, oncosurgeons and neurosurgeons need to be familiar with the clinical presentation and approach for IEES. 6
Location of Common Spinal Lesions.
Diagnosis of ES mainly depends on pathological evaluation. Histologically, the tumour cells show features of undifferentiated mesenchymal cells. On light microscopy, undifferentiated small tumour cells are predominantly seen with round, centrally placed, basophilic hyperchromatic nuclei, a diastase-digestible scant amount of clear cytoplasm with indistinct cellular borders. 15 Intracytoplasmic glycogen and neuronal differentiation may be present. 16 A biphasic population of dark and light is common. As seen in this case, pPNET sometimes specifically contain neuroblastic pseudo-rosettes called as Homer-Wright rosettes. 3
Cytoplasmic vacuoles may be seen in atypical ES. In atypical ES, tumour cells are larger than those in conventional ES with greater heterogeneity and may have focal spindle cell features. 17 These features were not seen in this case.
EES/pPNETs will show strong cell surface glycoprotein CD99 on immunohistochemistry, which is best demonstrated by MIC-2, antibodies O-13 or HBA-71. 11 It is positive in about 90% of EES/pPNET cases is one of the most accurate diagnostic markers. However, it is not exclusively specific for EES/pPNET. 18 Most EES/pPNETs are positive for Fli-1 and β2-microglobulin. About 25% of these cases demonstrate aberrant expression of epithelial marker-like keratin. Expression of a minimum of two neuroglial antigens, such as protein S100, neuron-specific enolase (NSE), synaptophysin or chromogranin, is essential to distinguish pPNET from ES, with the latter showing typically less neuronal differentiation. In 90% of ES/pPNETs, chimeric gene (EWSR1- FLI1) is found due to translocation t (11; 22) (q24; q12). This translocation in the nuclei of neoplastic cells can be detected by the FISH technique. 3
Treatment and prognosis: IEES are extremely rare malignant tumours, which in turn need multimodal therapy with surgical resection with a negative margin, craniospinal radiotherapy and systematic chemotherapy. 6 According to treatment guidelines, patients with ES receive neoadjuvant multiagent chemotherapy with vincristine, adriamycin and cyclophosphamide alternating with ifosfamide and etoposide upon diagnosis of ES by biopsy. 19 However, spinal tumours are usually diagnosed after resection. Complete resection of the lesion, the cuff of normal tissue and bone are required when feasible. Radical resection with removal of the entire epidural space may not be possible. Spinal primary lesions have a poor prognosis, mainly due to the difficulty of achieving a complete negative resection margin. 20 Postoperative adjuvant radiation is required to improve local control in cases of tumour spill at the time of surgery or if with positive surgical margins. 19 Many features define the prognosis and help direct the modality of treatment required. The prognostic factors include age, size and location of tumour, presence of distant metastases and presence of certain chromosomal translocations. 20 Additionally, larger tumours, fever, anaemia, raised serum LDH, a short interval between onset of symptoms, metastatic disease and diagnosis indicate worse prognosis. 21
Conclusion
ES/pPNETs presenting as intradural extramedullary location are an extremely rare entity and mimic more common primary spinal cord tumours. Therefore, neurosurgeons and oncologists need to be familiar with the clinical presentation and evaluation of IEES/PNETs as it warrants aggressive treatment modalities. Careful correlation of clinical presentation, cytomorphology, histopathology along with immunohistochemistry and molecular analysis is indispensable for the diagnosis of primary spinal ES/PNET from other primary spinal tumours.
Authors’ Contribution
PBK obtained details, analysed the data, wrote the manuscript and obtained the references. SD and MG were the physicians who treated the patient and contributed to the manuscript. HP contributed to the concept, design of the case report and structuring of review of literature. HP and DJ were the consultant pathologists who reported the cytology, histopathology and immunohistochemistry of the case, contributed to the overall manuscript supervision, review and guidance. All authors have approved the final version of the manuscript.
Statement of Ethics
Written informed consent was obtained from the patient for publication of this case report and any accompanying images.
Footnotes
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship and/or publication of this article.
Funding
The authors received no financial support for the research, authorship and/or publication of this article.