
Abstract
Objectives
To calculate design-corrected estimates of the effect of screening on prostate cancer mortality by centre in the European Randomised Study of Screening for Prostate Cancer (ERSPC).
Setting
The ERSPC has shown a 21% reduction in prostate cancer mortality in men invited to screening with follow-up truncated at 13 years. Centres either used pre-consent randomisation (effectiveness design) or post-consent randomisation (efficacy design).
Methods
In six centres (three effectiveness design, three efficacy design) with follow-up until the end of 2010, or maximum 13 years, the effect of screening was estimated as both effectiveness (mortality reduction in the target population) and efficacy (reduction in those actually screened).
Results
The overall crude prostate cancer mortality risk ratio in the intervention arm vs control arm for the six centres was 0.79 ranging from a 14% increase to a 38% reduction. The risk ratio was 0.85 in centres with effectiveness design and 0.73 in those with efficacy design. After correcting for design, overall efficacy was 27%, 24% in pre-consent and 29% in post-consent centres, ranging between a 12% increase and a 52% reduction.
Conclusion
The estimated overall effect of screening in attenders (efficacy) was a 27% reduction in prostate cancer mortality at 13 years’ follow-up. The variation in efficacy between centres was greater than the range in risk ratio without correction for design. The centre-specific variation in the mortality reduction could not be accounted for by the randomisation method.
Introduction
In the European Randomised Study of Screening for Prostate Cancer (ERSPC), randomisation method varied among centres. 1 In some centres, a target population was identified and randomly sampled (Finland) or allocated (Sweden, Italy, France) to the intervention (invitation to screening) or control arms of the trial. In other centres (the Netherlands, Spain, Belgium, and Switzerland), the target population was first invited to consent to participate, and only those consenting were randomised, to either the intervention (invited to screening) or control arm. These designs are called pre- and post-consent randomisation, or effectiveness and efficacy design, respectively. It has been suggested that randomisation methods may have introduced a bias in the published ERSPC results. 2
Efficacy is the effect on outcome in theoretically optimal conditions (e.g. with 100% compliance/attendance), while effectiveness is the effect on outcome in a real-life population setting. In screening, and other public health activities, the difference between these designs stems mainly from the extent of non-response. Attendance of those randomised to the intervention arm is generally higher with the efficacy design because the subjects have already indicated their willingness to take part. The attendance proportion is a major determinant of the impact of population screening on mortality outcomes. However, coverage, the proportion of those in the total target population who are screened, may be less in trials with a post-consent randomisation design than with a pre-consent design, because of the two phase process of both consenting and attending. There may also be differences in the underlying risk (of either all-cause or disease-specific mortality) in the randomised populations due to the ‘healthy volunteer’ effect, 3 although there is no evidence that this affects the relative risk due to the intervention.
The choice of design depends on both ethical and practical constraints. Some ethical review boards regard a study without consent of the controls as unethical (in which case only an efficacy study is possible), whereas some consider that, as the whole population is not covered by the trial (e.g. there may be restrictions by study area, calendar time, age, and other characteristics), design choice can be based on whichever provides data of more scientific value. Ethical review board views are also reflected in local legislation. In the ERSPC, the choice was in line with different national legal regulations.
The two designs serve different purposes. The post-consent randomisation (efficacy) design in prostate cancer screening addresses effect in those who choose to be or are actually screened, compared with a control group of men offered the normal health care practice, which will include opportunistic PSA-testing. 4 For brevity, we call this a clinical purpose, as it relates closely to the issue of clinical practice. The post-consent randomisation (effectiveness) design addresses the effect of a screening programme as public health policy in the target population, compared with normal clinical practice without a screening programme and therefore serves a public health purpose.
We previously reported a 21% reduction in prostate cancer mortality at 11 and 13 years of follow-up in men aged 55–69 invited to screening.5,6 This overall estimate did not take into consideration the two different designs of the included centres. Attendance for screening will tend to be lower with the pre-consent randomisation design, although even with post-consent randomisation there will be some non-attenders. After correction for non-attendance and adjustment for selection bias due to a likely higher mortality in non-attenders for screening, the overall efficacy was estimated at 27% at 13 years of follow-up. However, large differences in the uncorrected prostate cancer mortality reduction between centres were observed, from a 14% increase (Switzerland) to a 38% reduction (Sweden).5,6
The reason for the differences in effect between the centres is likely to be multifactorical. In this paper, we correct only for the design of effectiveness or efficacy. We also discuss the implications of these two different study purposes on the design and analysis of a screening study. We report the design-corrected efficacy and the effectiveness of screening for prostate cancer in the ERSPC screening trial by centre, with follow-up until 31 December 2010, censored at 13 years, and address the question of variation in effect between centres that can be accounted for by the different designs.
Methods
Population
Number of men in the target population and screening arm, years of intake, and mean years of follow-up by centre in ERSPC.
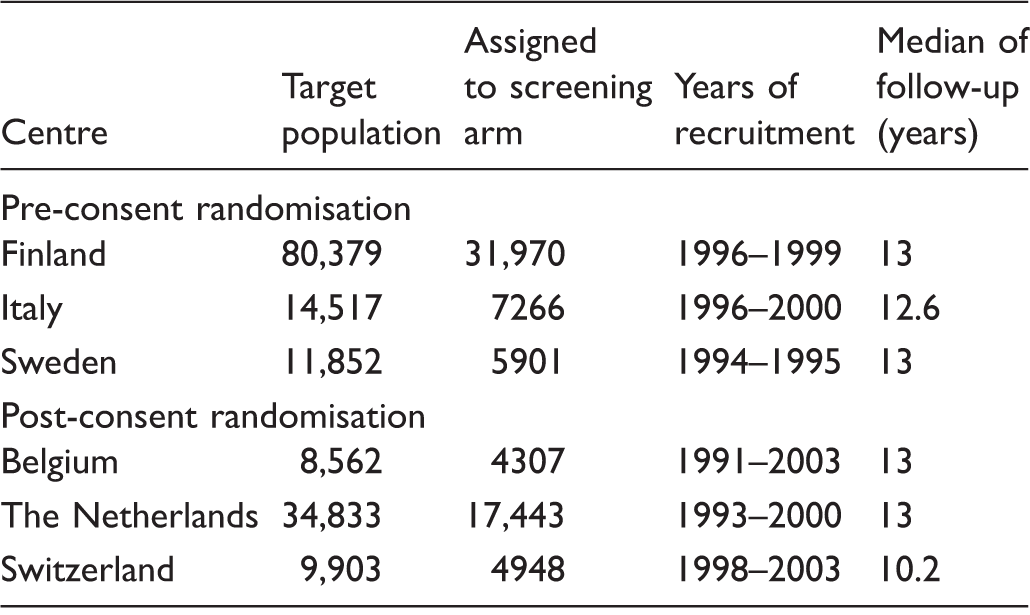
Note: Core age group 55–69, follow-up to 31 December 2010, censored at 13 years.
Definitions and notations
We define the outcome as death from prostate cancer, and attendance as attendance in response to first invitation to screening.
We use the following notations: M(p) = mortality from prostate cancer in the whole target population; for the post-consent randomization (efficacy) design, this includes the population from whom men were recruited, which is generally not known. M(v) = mortality from prostate cancer in the men consenting to take part (post-consent randomization (efficacy) design) M(a) = mortality from prostate cancer in the attendees to screening M(na) = mortality from prostate cancer in non-attendees (among invitees to screening) α = person years in attendees as a proportion of the person years in the invited target population (pre-consent randomization (effectiveness) design) γ = person years in attendees as a proportion of the person years in those consenting and randomized to the intervention arm (post-consent randomization (efficacy) design)
Invited are those randomized to the intervention arm in the total target population (effectiveness design) or in the consenters (post-consent randomization (efficacy) design).
We further denote:
M0(.) = prostate cancer mortality assuming no screening offered
M1(.) = prostate cancer mortality assuming screening offered
For each of. = p, v, a and na.
The basic relations, that link the quantities above, are

These relations provide estimates of the mortality rate in the attenders in the absence of screening, by subtracting from the mortality in the control arm the mortality equivalent to that in the non-attenders in the intervention arm, and thus take account of selection bias.
With these denotations, we can define


Estimation of effectiveness
The pre-consent randomisation design provides a direct estimate of effectiveness. In equation (3), M1(p) is the prostate cancer mortality in the total (invited) intervention arm and M0(p) is the mortality from prostate cancer in the control arm. Both quantities are known from the data. The post-consent randomisation design does not provide data on effectiveness. To estimate effectiveness would require the person years in the consenters as a proportion of those in the total target population, together with the mortality in non-consenters, to be known, in addition to the trial data themselves. This information is rarely available, and was not available in all ERSPC centres. More importantly, the inclusion of a two-phase screening process both consenting and attending means that such an estimate would lack real life applicability. In real life, only a single phase will exist: that of attending, or responding to the invitation. In an efficacy trial, the sum of non-consenters and non-attenders will differ from the number of non-attenders in an effectiveness trial because of the difference in motivation. Conceptually, to estimate the effectiveness from an efficacy trial requires restrictive assumptions, and we do not present any such estimates for the pre-consent centres in the ERSPC trial.
Estimation of efficacy
Transformation in the pre-consent randomisation (effectiveness) design to the efficacy E(a) takes place with the basic relation (1) that has been described elsewhere,
8
and which takes account of selection bias due to differential mortality in non-attenders, as well as the dilution due to non-attendance itself.
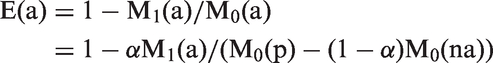
Here M0(na) is the mortality in those randomised in the intervention arm who did not attend. M1(a) is the mortality in attenders, i.e. in those actually screened. M0(p) is the mortality in the control arm, and α is the person year proportion of attenders in the screening arm. All these quantities are directly estimable from the data.
Even with the post-consent randomisation design, some correction is necessary to produce an estimate of efficacy with 100% attendance, because not all of those who consented and were randomised to the intervention arm actually attended, and some selection bias may still be present. The expected mortality in those attending can be estimated in a similar way to the pre-consent randomisation design, using the basic relation in the consenters (2) between the risk of death among non-attenders and controls. Simple arithmetic yields
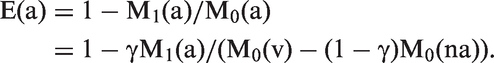
Here M1(a) is the mortality among those screened (the attenders), M0(v) is the mortality in the control arm of those consenting, M0(na) is the mortality in the consenters in the intervention arm who did not attend, and γ is the person year proportion of attenders in intervention arm. All these components are known and estimable from the data.
Results
Number of men, person years, and number of prostate cancer deaths by arm, attendance status, and centre in ERSPC.
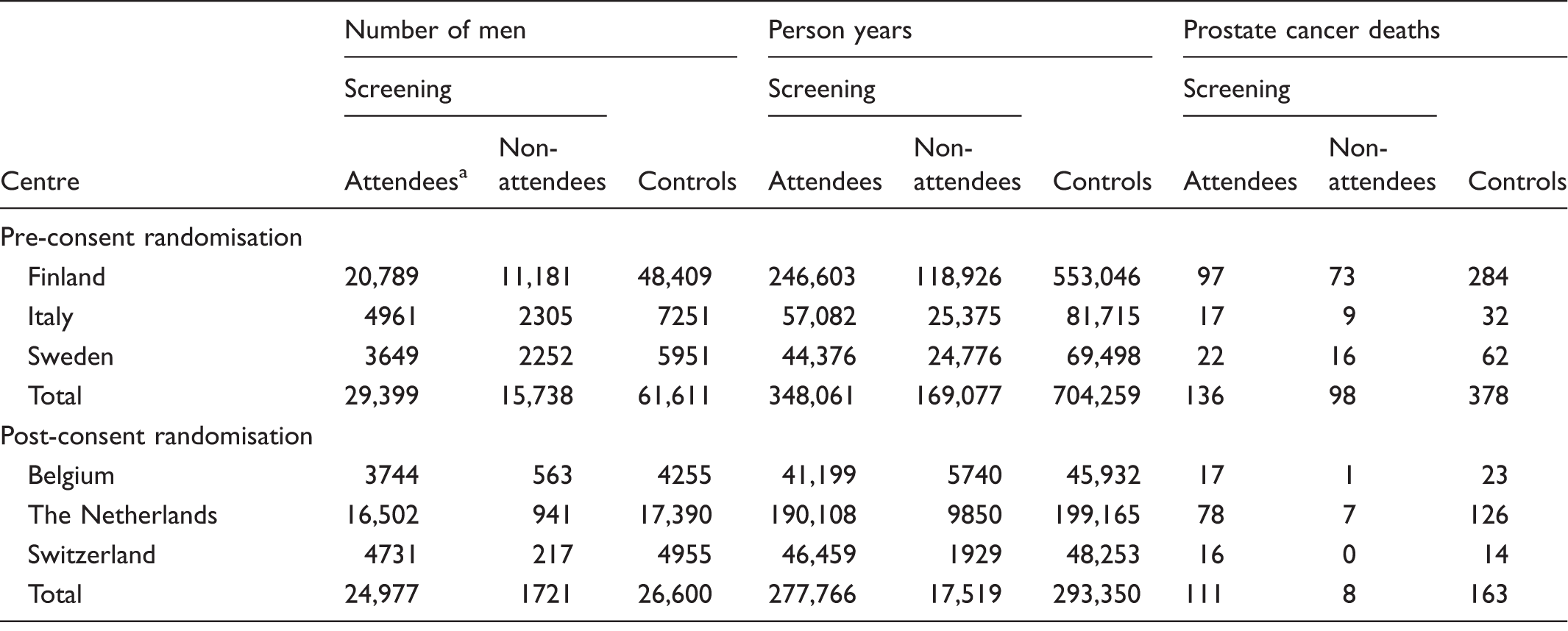
Note: Core age group 55-69, follow-up to 31 December 2010, censored at 13 years.
Responders to the first invitation.
Effectiveness (in the population) and efficacy (in attenders) and by ERSPC centre and arm.
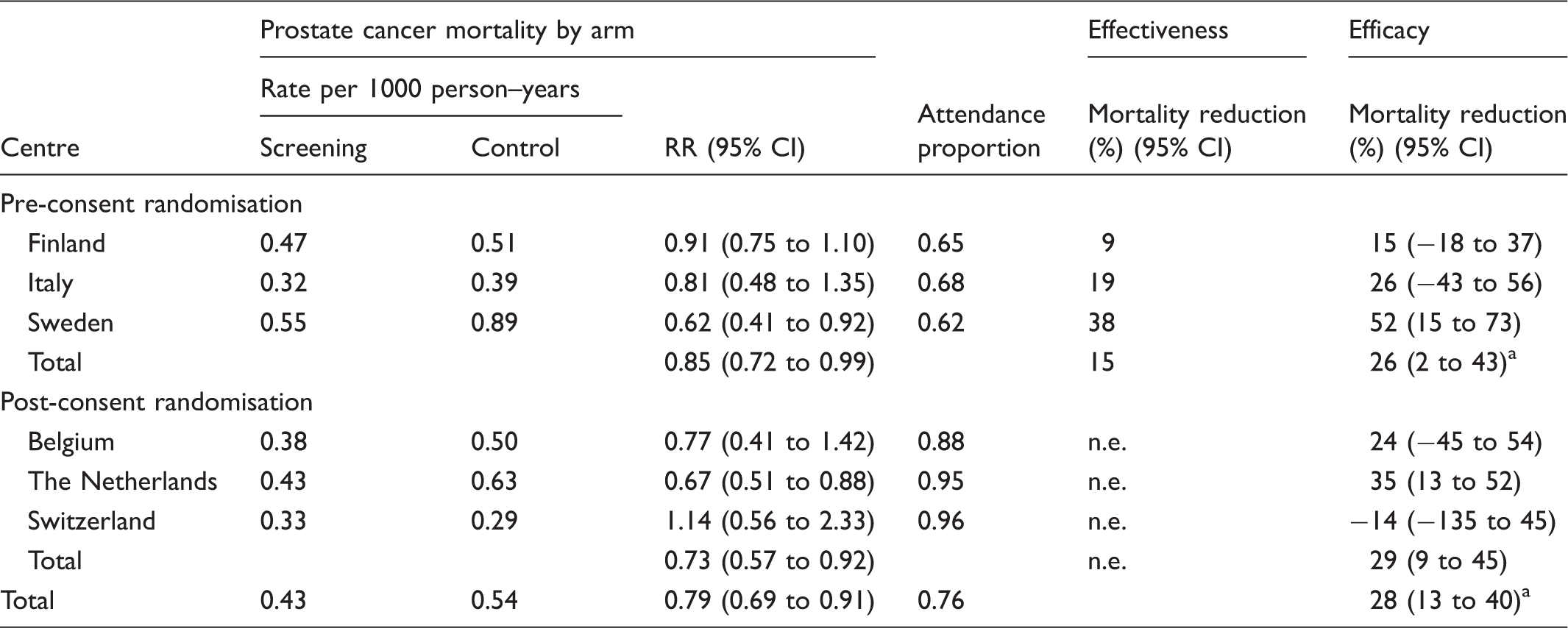
Note: Core age group 55–69, follow-up until 31 December 2010, censored at 13 years. n.e., not estimable.
With adjustment for the control population in Finland.
For estimates of efficacy in attenders, the overall risk ratio was 0.72 (95% CI 0.60–0.87), and the efficacy (1-RR)×100 increased to 28%. It was smaller in centres with pre-consent randomisation design (26%) than in those with post-consent randomisation design (29%). The range of 1-RR was 0.66 (from 0.52 to −0.14).
Discussion
We calculated adjusted estimates of mortality reduction for the ERSPC centres to improve comparability between centres. Randomisation in the centres was by two different methods, post-consent randomisation in Belgium, the Netherlands, and Switzerland, and pre-consent randomisation in Finland, Italy, and Sweden. In Italy and Sweden, a random allocation in 1:1 ratio was followed, whereas in Finland 32,000 of more than 80,000 men were randomly sampled to the screening arm. It has been suggested that the randomisation methods may have introduced a bias 2 and resulted in too large an estimated effect with pre-consent randomisation, 9 and that therefore the pooling of ERSPC centres may be inappropriate. 10 While the purpose of randomisation per se is to remove bias, application of different randomised designs may cause incomparability. In the present study, we correct for the incomparability, and relate the randomisation method to the effect in those actually screened or in the target population (i.e. to the purpose of the trial). The correction for efficacy had a greater impact in centres with a pre-consent randomisation (effectiveness) design than in those with a post-consent randomisation (efficacy) design.
The different designs correspond to different contexts of screening. In practice, both designs compare an organised screening programme with routine clinical practice, which will include opportunistic screening. Opportunistic or spontaneous PSA-testing, in either the intervention or control arm, is called contamination. The performance of the test in the absence of such spontaneous use is difficult to measure once a test is approved, but any attempt to correct for contamination methodologically will have the potential for bias. 8 With post-consent randomisation, knowledge of the randomisation may affect the probability of having a spontaneous test in those allocated to the control group, resulting in more treatment, and possibly an effect on mortality, but in a non-measurable way. It is, therefore, possible that the efficacy design underestimates the effect in those actually screened. In the effectiveness design, where individuals in the control arm are not contacted, the randomised study itself is less likely to affect the PSA-testing in the controls. We have not made such an assumption-based correction in this study.
Post-consent randomisation is specifically designed to provide an estimate of efficacy; however, the relative risk of prostate cancer death between the arms should still be corrected for the non-attendance in those consenting. Pre-consent randomisation is designed to estimate effectiveness in the target population, but at the same time it provides an estimate of efficacy. Therefore, any changes related to the screening (exposure) and to the treatment and, hence, to death, are likely to be more comparable with the population at large in the effectiveness trial than in the efficacy one. Furthermore, it is difficult to see how the exposure to any medical services in a randomised trial that is identical in the controls and in the population at large would violate any ethical rules.
The Prostate, Lung, Colorectal and Ovarian (PLCO) cancer screening trial conducted in the USA enrolled over 150,000 subjects at 10 different screening centres, some of which used a ‘single consent’ process (post-consent randomisation), and some a ‘dual consent’ process, where randomisation was carried out after initial consent to follow-up, and subjects randomised to the intervention arm were asked to consent again to screening. 11 The odds ratio of non-compliance was 2.2 in the dual consent centres, even after adjustment for other factors. Contamination by screening in the control arm was a major issue in the prostate screening trial in PLCO, 12 but data on contamination according to the consent process have not been published.
We believe that, from a scientific point of view, the pre-consent randomised design without explicitly consenting the controls is superior to the post-consent randomised design because, as demonstrated above, the former can be used to provide results on both the clinical problem of efficacy and on the public health question of effectiveness, whereas the latter provides results only on efficacy. However, the method above only provides adjusted estimates of efficacy in those accepting the first invitation to screening, and more sophisticated methods are required to study the effect of different patterns of subsequent screening attendance.
Even after correcting for the differences in design by estimation of efficacy, considerable variation remained between centres. As discussed elsewhere, possible reasons for this variation include differences in the extent of contamination by PSA screening in the control group, and variations in screening protocol, including the number of screens, and the length of the screening interval. 6
Efficacy was estimable in all ERSPC centres, with minor restrictive assumptions. After correction for non-attendance and selection bias, the overall efficacy (effect in attenders) was a 28% reduction in prostate cancer mortality. The effect estimate in the ERSPC of 21% in men invited 6 was a mixture of effectiveness and efficacy. Efficacy (effect in attenders) was larger in centres with post-consent randomisation than in those with pre-consent randomisation design, but the difference in the overall estimate of efficacy between the two groups of centres was substantially smaller than that in the crude estimate of relative mortality risks. However, the correction for study design did not reduce the variation between individual centres, suggesting that centre-specific variation in the mortality reduction could not be accounted for by the randomisation method.
Footnotes
Declaration of conflicting interests
The author(s) declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: Dr Stenman declares the following conflicts of Interest: Co-holder of patent for free PSA. Consulting for PerkinElmer-Wallac, Abbott Diagnostics, Orion Diagnostics.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: Funding/Support and role of the sponsor: European Randomized Study of Screening for Prostate Cancer. Dr Sigrid Carlsson’s work on this paper was supported in part by a Cancer Center Support Grant from the National Cancer Institute made to Memorial Sloan Kettering Cancer Center (P30 CA008748). Dr Carlsson is also supported by a post-doctoral grant from AFA Insurance.