
Abstract
Objectives
In Guadeloupe, an island in the French West Indies, a universal newborn screening programme for sickle cell disease and other abnormal haemoglobins was initiated in 1984. In 1990, a comprehensive sickle cell centre was established to carry on the management programme. We here report the main results from the newborn screening programme from 1984 to 2010, and consider how the establishment of the sickle cell centre affected the programme.
Methods
All blood samples were screened for the haemoglobinopathies using two reference methods in a single reference diagnosis laboratory. DNA analyses were also performed for confirmatory tests and analysis of the globin gene status.
Results
Between 1 January 1984 and 31 December 2010, 178,428 newborns were screened at birth, and 585 children were diagnosed with major sickle cell syndromes (ie. an overall incidence of 1 in 304 births). Sickle cell anaemia (haemoglobin SS disease) was the most frequently observed (1 in 575 births), followed by haemoglobin SC disease (1 in 771 births) and haemoglobin Sβ-thalassemia disease (1 in 4,243 births). Some other rare haemoglobin variants were also detected, the most common being HbDPunjab. The establishment of a comprehensive sickle cell centre resulted in a significant improvement in the screening coverage (p < 0.001) and a significant reduction of the delay between diagnosis and the first medical visit (p < 0.001).
Conclusion
The universal screening programme has made it possible to establish the incidence of the major sickle cell syndromes in Guadeloupe, and the management centre has improved its efficiency.
Introduction
Sickle cell disease (SCD) encompasses a group of autosomal recessive disorders, characterized by the production of high levels of the abnormal sickle haemoglobin (HbS). These disorders share several clinical features including severe haemolytic anaemia, painful vasoocclusive crises, susceptibility to serious infections, stroke, and chronic damage to the lungs, bones and kidneys. The most common type of sickle cell disease is the homozygous condition known as haemoglobin SS disease or sickle cell anaemia. Other sickling diseases occur when the gene coding for HbS (βS gene) inherited from one parent is associated with either the βC gene which encodes for another abnormal haemoglobin variant (HbC), or a beta-thalassemia (β-thal) gene, resulting in conditions known as sickle cell-haemoglobin C disease (SC) and sickle beta-thalassemia (Sβ-thalassemia) syndromes, respectively. Other less frequently encountered β-globin variants associated with the βS gene such as βDPunjab or βOArab or variants with a dominant expression, carried at the heterozygote state, such as βSAntilles or βSOman may also lead to the clinical expression of the disease. 1
The purpose of newborn screening is to facilitate the early identification of a disease so that an intervention that significantly improves the natural history of the disease might be instituted before significant morbidity or early mortality occurs. For sickle cell disease, newborn screening mainly allows early penicillin prophylaxis of pneumococal infections, aggressive vigilance for routine infant immunizations and pneumococcal vaccination at age two, and preventive parental education prior to the appearance of the first symptoms of the disease. 2 Gaston et al showed a drastic reduction of mortality in sickle cell infants due to peniciloprophylaxis, 3 and other studies have clearly demonstrated the benefit of newborn screening on sickle cell disease related mortality.4–6
Guadeloupe is a French archipelago comprising a main group of six closely situated islands (Basse-Terre, Grande-Terre, Marie-Galante, La Desirade, Terre-de-Haut, Terre-de-Bas) and two Northern Islands 250 km away (Saint-Martin, Saint-Barthelemy). Since 2003, Saint-Martin and Saint-Barthelemy have been autonomous administrative collectivities.
In 1984, a universal newborn screening programme for SCD and other abnormal haemoglobins was initiated in Guadeloupe. Initial data from this programme confirmed that SCD is the most frequent inherited disease in the Guadeloupian population. 7 In 1990, a comprehensive management programme including medical, psychological and social management of patients, extensive carrier screening, genetic counselling and prenatal diagnosis was implemented. The newborn screening programme is part of the French national programme for the newborn screening of SCD and was supported by the French Association for Screening and Prevention of Child Handicaps and by the national public medical insurance system for salaried persons.
We here report the results of the programme from 1984 to 2010. These results establish the incidence of SCD at birth in the Guadeloupian population, the nature and frequency of the most prevalent haemoglobin variants, and of the alpha- and beta-thalassemia determinants. We also describe the impact the establishment of a centralized comprehensive SCD centre has had on the newborn screening coverage and the delay between diagnosis and enrolment in an adequate medical management programme.
Methods
Sampling collection and screening procedure
From 1984 newborn blood samples were routinely collected from delivery sites in the Guadeloupe archipelago. For organizational reasons, up to 1989, the collection was quite difficult in Saint-Martin island. Samples were sent to the reference diagnosis laboratory initially located in the facilities of the University Hospital of Pointe-à-Pitre (1984 to 1991), and then at the comprehensive sickle cell centre.
Before 1995, diagnosis was performed on cord blood samples collected on the day of birth and afterwards on dried blood spots taken two days after birth. Diagnosis date was therefore defined as the birthdate. Until 1991, primary screening was based on thin layer isoelectric focusing (PerkinElmer, Massachusetts, USA) and haemoglobin citrate agar electrophoresis as a confirmatory test. After this confirmatory testing was conducted by high performance liquid chromatography (HPLC, BioRad Laboratories, Hercules, CA) which allows the quantification of the different haemoglobin fractions according to the national guideline for laboratory procedures.8–11
From 1984 to 1990, the Mother and Child Unit of the department political authorities (Conseil Général) of Guadeloupe provided information to parents which allowed diagnostic test for newborns with positive results on primary screening. At that time, retrospective data were collected from the places of birth and Mother and Child unit files and children were progressively included in a comprehensive care programme managed by the paediatric departments of the local hospitals. Since 1990, the nurse coordinator of the comprehensive sickle cell centre has visited the parents of infants with a positive primary screening result, at home, as soon as possible, for information and confirmatory testing.
The diagnostic test consists of haemoglobin analysis of both parents. When results are concordant, the first medical visit is scheduled and the preventive care starts (penicillin prophylaxis, vaccinations, and parental education). If the results from the parents are discordant with the newborn primary screening result or not available for both of them, confirmatory testing is performed on the newborn by phenotypic analysis. In addition to handling SCD cases and their families, a letter of information is sent by the sickle cell centre to the families of all carriers (AS, AC and other), in which a familial haemoglobin screening is suggested.
Molecular analysis
Over time, various genotyping methods, including reverse dot blot, PCR-RFLP, and direct DNA sequencing, were used to identify beta-thalassemia mutations, to identify uncharacterized Hb variants and to resolve ambiguous primary screening results as previously described.12,13 DNA analysis was performed for confirmatory diagnosis if the primary screening result was FSD phenotype (haemoglobin sickle D disease) and if a blood transfusion was administered before screening. The alpha-globin gene status was determined by GAP-PCR for the SCD children as previously described;14,15 alpha-globin gene status was not characterized for children with FAD (haemoglobin D trait) phenotype. Hereditary persistence of fetal haemoglobin (HPFH) alleles were characterized using multiplex GAP-PCR as previously described.16,17
Data collection
All neonatal screening data (haemoglobin phenotype at birth, genotypic or phenotypic confirmed diagnosis, blood haemoglobins levels, globin genes status) were prospectively collected in the laboratory diagnostic database.
The date of the first medical visit was recorded from the medical files. The medical management delay time was defined as the delay between the birthdate and the date of the first medical visit (meaning the age of the infant at first medical visit).
The whole number of births per year was obtained from the Mother and Child unit according to the registers at delivery places. The neonatal screening coverage was defined as the number of babies tested, over the absolute number of births.
Statistics
Allele frequencies were estimated by gene-counting. Comparison between the observed frequencies of SS and of SC disease (which deviated from that predicted by the Hardy-Weinberg equilibrium) was tested by Pearson’s chi-squared test. The qualitative variables were expressed as median and range and compared using the Mann-Whitney test.
Results
Between 1 January 1984 and 31 December 2010, 178,428 newborns were screened at birth for sickle cell disease in Guadeloupe, including Saint-Martin and Saint-Barthelemy. Among the screened samples, we failed to obtain definitive results in 239 (0.13%) cases owing to lack of a repeat samples, due either to the death of the child or loss to follow-up. Of the 239 cases, 213 were heterozygous for a variant other than those which could be identified by initial screening, such as haemoglobin S, haemoglobin C or haemoglobin D. Two cases were double heterozygote for haemoglobin S and an uncharacterized variant. Definitive diagnosis was also not obtained for 7 cases with initial results of FS and 17 with FC phenotype.
Incidence of homozygotes, compound heterozygotes and simple heterozygotes for haemoglobin variants.
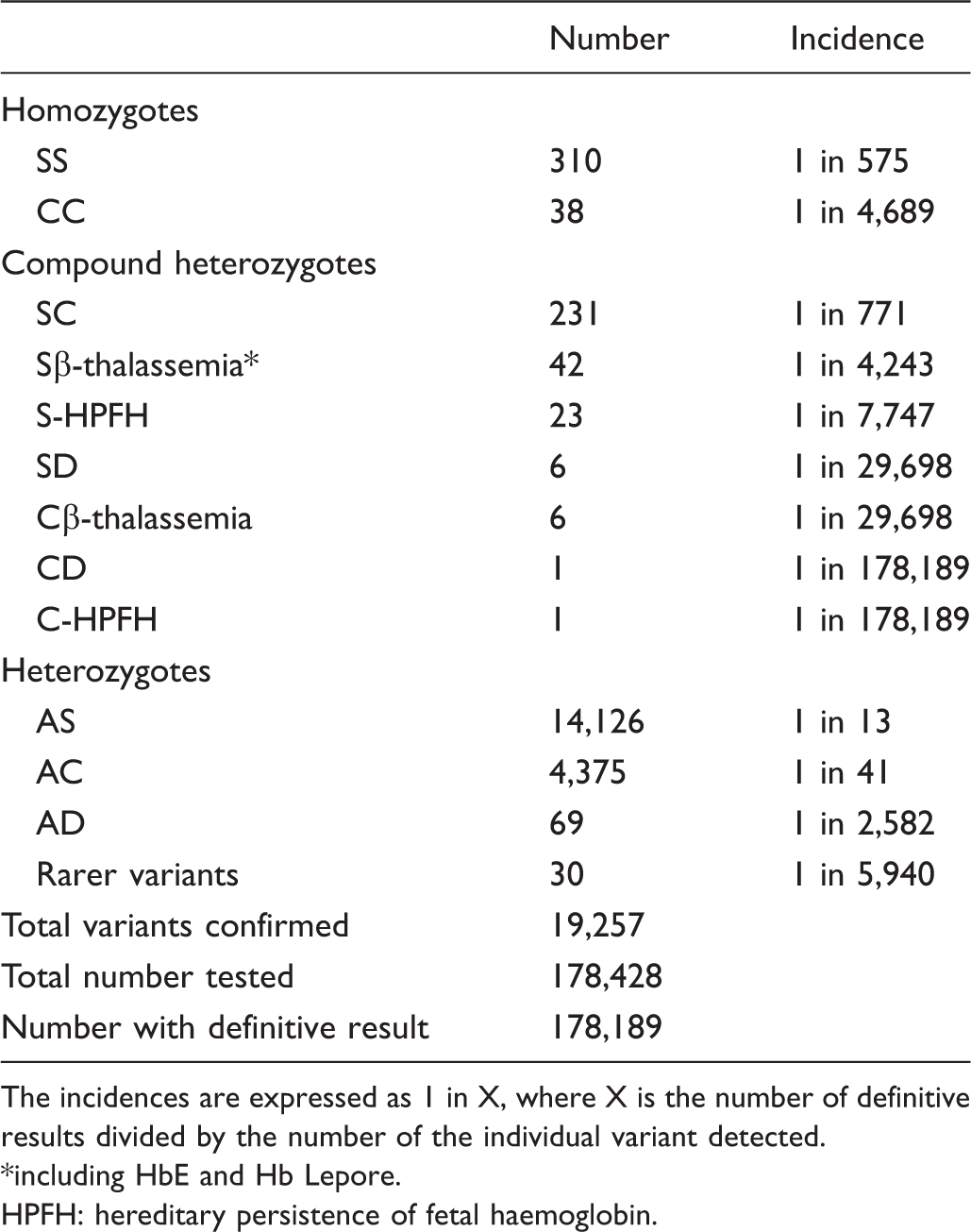
The incidences are expressed as 1 in X, where X is the number of definitive results divided by the number of the individual variant detected.
including HbE and Hb Lepore.
HPFH: hereditary persistence of fetal haemoglobin.
Allele frequency of the clinically significant beta globin variants.
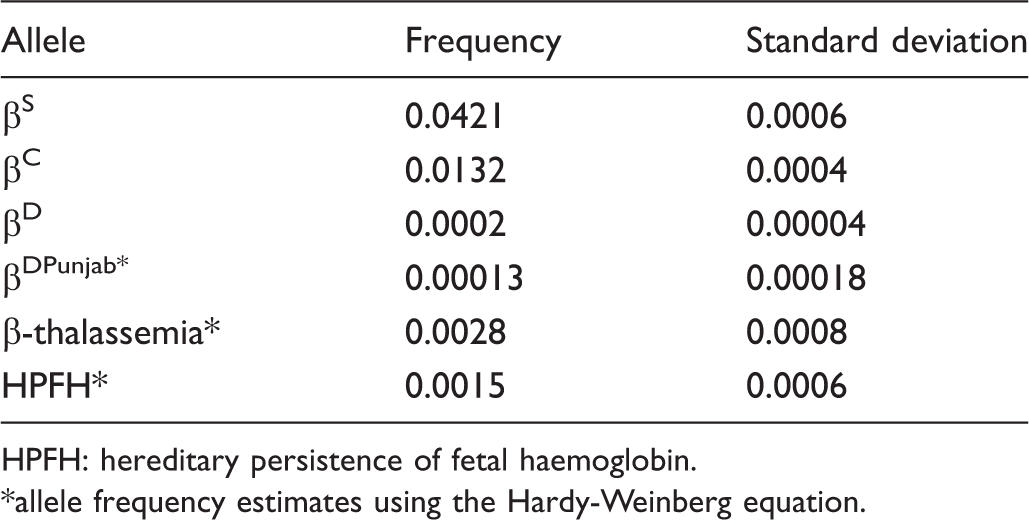
HPFH: hereditary persistence of fetal haemoglobin.
allele frequency estimates using the Hardy-Weinberg equation.
The observed frequency of the major genotypes deviated from that expected from the Hardy-Weinberg equation (χ2 = 8.38, df = 3, p < 0.04), 65.6% of the deviation being accounted by the excess of SC genotype and 18.8% by the apparent excess of CC cases (data not shown).
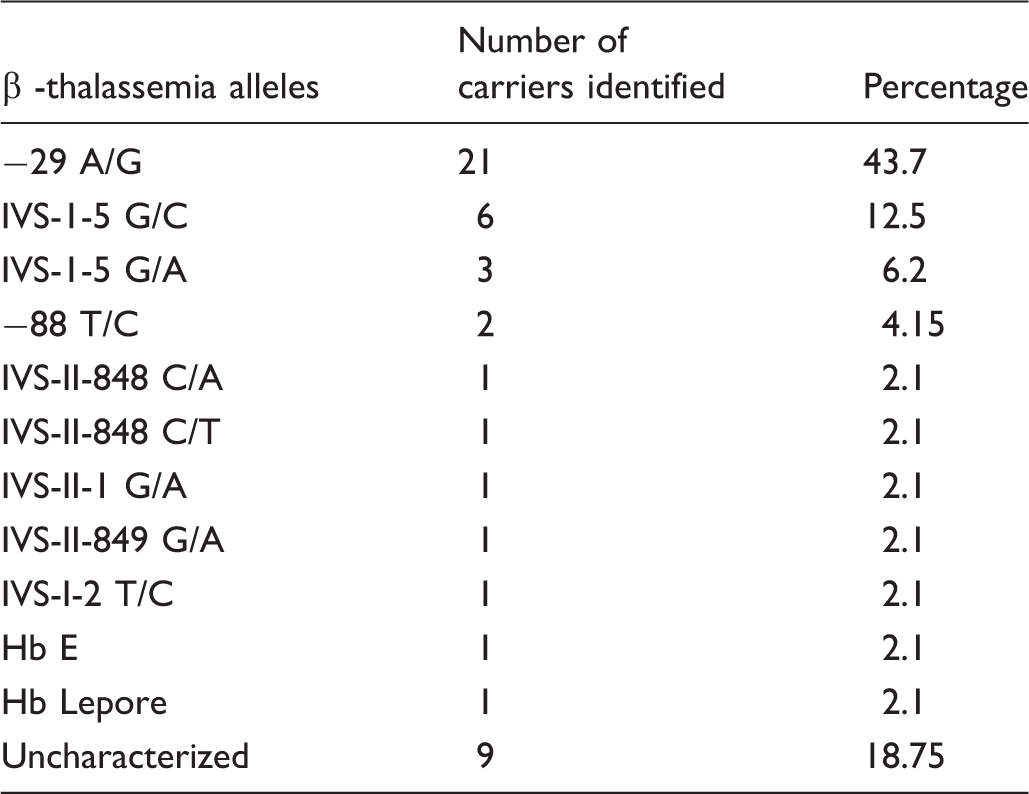
β thalassemia variants identified.
Alpha globin genes status of neonatal screened children with sickle cell disease in Guadeloupe.
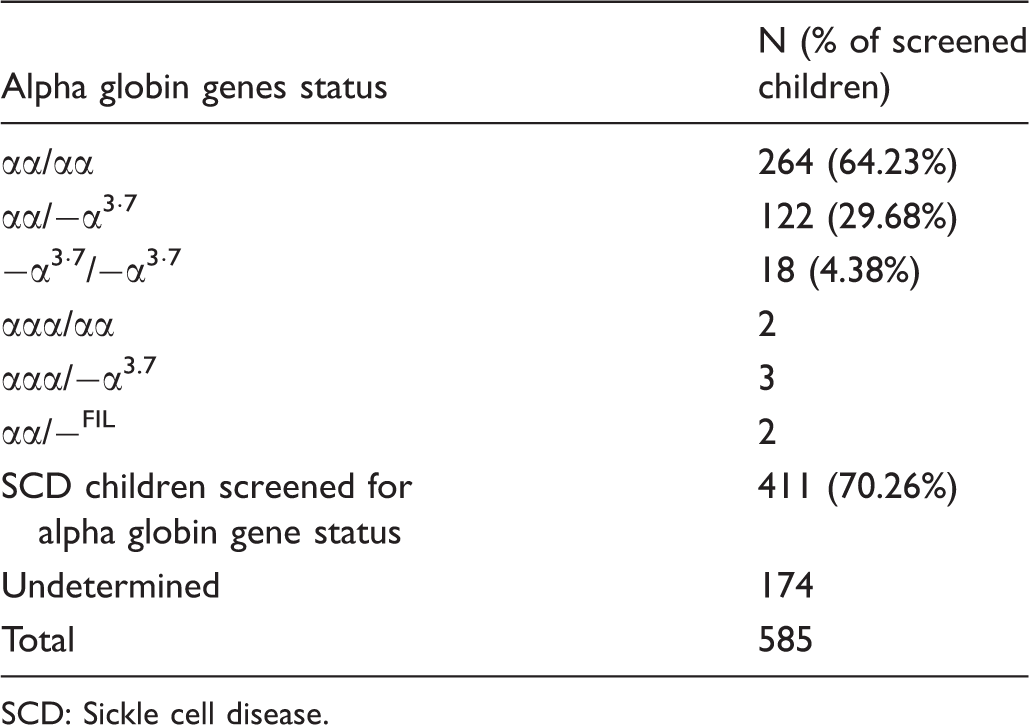
SCD: Sickle cell disease.
The neonatal screening coverage for the whole population and the overall medical management delay time for the subset of 452 children who attended the comprehensive sickle cell centre have been evaluated over the whole study period. Periods before and after the setting-up of comprehensive sickle cell centre in 1990 were compared, and showed a significant improvement in the screening coverage (83.02% [range: 72.5–90.6] vs 97.61% [range: 90.4–99.8], p < 0.001). Figure 1 shows a significant reduction of the delay time for the latter period (p < 0.001).
Evolution of mean medical management delay time of SCD infants from 1984 to 2010.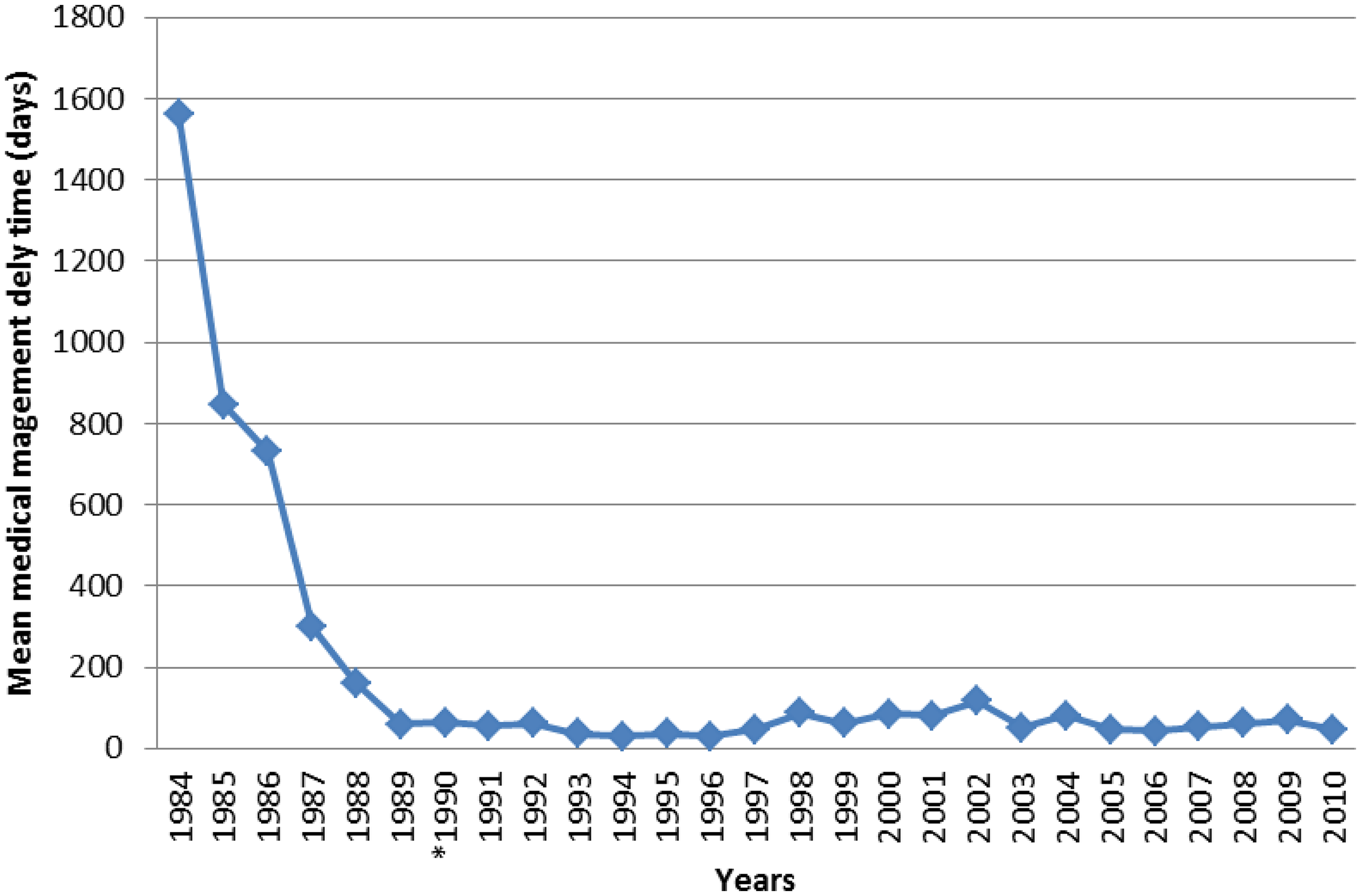
Discussion
This study, based on the results of the universal newborn screening (UNBS) programme for sickle cell disease over a 27 year period, provides the most objective information on the frequencies and incidences of SCD syndromes in Guadeloupe. Furthermore, the establishment of a sickle cell centre seven years after the beginning of the programme enables evaluation of the impact of a centralized organization on two main areas of concern in an SCD newborn screening programme, namely the coverage of newborn screening, and the delay between the identification of an affected child and medical follow-up.
From 1984 to 2010, 94% of newborns in Guadeloupe were screened for SCD. SCD (with various genotypes) was detected in 585 newborns. The most frequent syndrome is SS disease (53%), followed by SC disease (39.5%), and Sβ-thalassemia (7.2%). HbS and HbC were the most frequently encountered abnormal haemoglobins, in agreement with preliminary data previously published, 18 with an allele frequencies of 0.042 and 0.013 respectively.
Although β-thalassaemia major was not detected during this study, eleven different β-thalassaemia alleles were characterized. All these alleles had been already reported in the Guadeloupean population, the three most prevalent alleles (-29A/G, IVS-I-5 G/C and IVS-I-5 G/A) being described as frequent β-thalassaemia mutations in this population. 19 α-thalassemia trait was found at a high frequency, similar to that observed in individuals of African descent1,20 and in agreement with our previous results. 12 During this study, we identified a rare α-thalassaemia variant, namely –FIL, which had never before been detected in the Guadeloupean population and which accounted for 1.3% of the α-thalassemia alleles detected. No Haemoglobin H disease was identified during the course of this screening programme. Among the other abnormal haemoglobin variants which may lead to SCD syndrome, we detected Hb DPunjab, and two HPFH alleles known to be present in individuals of African descent, namely HPFH1 and HPFH2, were also detected.
Based on the genotype numbers calculated from the allele frequencies of the βA, βS and βC, there was an excess of patients with SC disease. The most likely explanation is an underestimation of the βC allele frequency, because confirmation tests were not obtained for 17 of 56 cases with FC phenotype. Similarly, seven of 340 children with FS phenotype were not fully characterized.
Overall, these limitations do not change the main findings of this report, which show that sickle cell disease is the most frequent genetic disease encountered in the Guadeloupean population 21 with a prevalence very similar of those detected in Jamaican and African-American populations (ie. three populations sharing African sub-Saharan origins).
The success of a newborn screening programme for any disease depends not only on the number of infants diagnosed, but on the timely fashion in which these children receive appropriate medical care. From 1984 to 1990 the UNBS was managed by several paediatric services; after 1990 the UNBS was included in a comprehensive management programme for sickle cell disease coordinated by the comprehensive sickle cell centre of Guadeloupe.
The first contact with the parents of an affected child was established by the nurse coordinator. After diagnosis, the affected children were followed up by the paediatrician in a dedicated hospital department (Sickle cell centre of the University Hospital of Pointe-à-Pitre, paediatrics unit of Hospital of Basse-Terre or paediatrics unit of the hospital of Saint-Martin) according to the French medical guidelines for sickle cell patients. Whatever their haemoglobin type, as soon as a sickle cell newborn is identified through newborn screening, the nurse coordinator introduces the child to the management programme. Medical teams pay special attention to SS and Sβ0thalassemia patients who have the most severe genotypes.
This system of organization avoids several limits reported in previous studies, where affected infants were managed by physicians not prepared for the follow up care of SCD children.22,23 The establishment of the comprehensive sickle cell centre in 1990 had a positive impact on both the percentage of newborns screened, which was higher than 98.5% since 2002, and delay in the medical management of sickle cell infants, with a mean medical management delay time of less than 2 months. Before the opening of the comprehensive sickle cell centre, newborns were screened at birth but systematic early medical management was not yet organized. For some children, the beginning of the medical follow up was very late, mainly after the occurrence of specific complications. This explains the long management delay time recorded before 1990. From 1989, the nurse coordinator began to refer the SCD infants early after screening to paediatric departments. Such early-age inclusion in the medical follow up had been shown to be important in sickle cell disease, as during early childhood years these infants are at high risk. Data from the Jamaica Sickle Cell Cohort Study show that approximately 14% of the SCD children died in the first two years of life before early-age interventions had been implemented, while 32% and 61% of children with haemoglobin SS disease became symptomatic by age of one year and two years respectively.24,25 Such improvements in the screening coverage rate and early inclusion of affected children were a direct consequence of the centralized management of the UNBS programme, with close relationships between the reference diagnosis laboratory and the sickle cell centre (the laboratory was a unit of the sickle cell centre until 2011; now both are located in the same building). This allows earlier pathology results to be available to the nurse coordinator, and ensures timely enrollment of the affected children. This experience confirms that a regional organization associated with a national approach allows a more efficient use of resources and ensures consistency of information dedicated to families and healthcare practitioners. 26
In our experience, the obstacles for UNBS were the early discharge of mothers after delivery but before their babies were screened, misinformation of newly enrolled maternity professionals, or use of wrong procedures for sending dried blood spots to the diagnosis laboratory. The main obstacles for longer delay for medical management were frequent relocation of migrant people, wrong address on blotting papers, and social difficulties.
Conclusion
The universal newborn screening programme conducted in Guadeloupe has enabled the estimation of the incidence rates for the major sickle cell syndromes as well as the frequencies of the most commonly encountered abnormal variants in this population. In addition, the management of the UNBS programme by an unique structure improved the efficiency of the programme in Guadeloupe.
Footnotes
Acknowledgements
The authors are grateful to Marie-Claire JEROLON for fully participating in this study, Professor Jacques ELION for careful reading of the manuscript and helpful discussion.
Funding
This research received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.