
Abstract
Three wholly aromatic polyamide–hydrazides I–III containing various amounts of meta-/para-phenylene rings and their multiwalled carbon nanotube (MWCNT) composites have successfully been synthesized. These polymers were synthesized via a low-temperature solution polycondensation reaction of either meta-aminobenzhydrazide or para-aminobenzhydrazide (PABH) with an equimolar amount of isophthaloyl chloride (ICl) producing polymer I (100% meta-phenylene rings) and II (50% para-phenylene rings/50% meta-phenylene rings), respectively. The polymer III (75% para-phenylene rings/25% meta-phenylene rings) was synthesized by reacting PABH with mixture of equimolar ratios of terephthaloyl chloride and ICl. MWCNTs (0.1 wt%) have been incorporated into these three polymer matrices via a solution sonication technique. The structures of the polymers were confirmed by elemental analyses, Fourier-transform infrared, and proton nuclear magnetic resonance spectroscopy. The homogeneous distribution of the MWCNTs within the polymer matrices was proved by field emission scanning electron microscopy, transmission electron microscopy, and atomic force microscopy observations. Thermal and optical properties of the as-prepared polymers were being enhanced to some extent upon inclusion of MWCNTs as illustrated from the results of thermogravimetric analysis and ultraviolet–visible spectroscopy.
Keywords
Introduction
Wholly aromatic polyamide–hydrazides are one of the most important classes of high-performance polymers. 1 -5 The repeating units of these polymers possess one amide (–NHCO–) and one hydrazide (–CONHNHCO–) linking bond between appropriate aromatic nuclei. A general structural formula of these polymers could be represented as follows:
–(–CO–NH–Ar–CO–NH–NH–CO–Ar′–(– n , where Ar and Ar′ most often stand for para-phenylene and/or meta-phenylene aromatic nuclei. These polymers are known to resist elevated temperatures in inert as well as oxidative atmospheres. They possess a good combination of high thermal stability of both the aromatic polyamides and the aromatic polyhydrazides. Their thermal aging and degradation behaviors have been the object of extensive study by many investigators. 6 -9 The solutions of this class of polymers in dimethylacetamide and dimethyl sulfoxide result in films and fibers with good rheological properties. Their drawn fibers possess high strength that exceeded those reported for glass fibers and steel wires. 10 Some examples of their technological applications are in tire-cords, impact-absorbing devices, fabrics, and ballistic protection.
The general method adopted to synthesize these polymers is a low-temperature solution polycondensation reaction of aromatic aminohydrazides with aromatic dicarboxylic acid dichlorides in an amide solvent. 11 -14 Completely ordered polyamide–hydrazides were prepared from diamines containing preformed hydrazides with diacid dichlorides. 15 A few polyamide–hydrazides which were obtained from dihydrazides having preformed amide linkages with diacid dichlorides have been reported. 16
Carbon nanotubes (CNTs) have long been recognized as the stiffest and strongest man-made material known to date. In addition, their high electrical conductivity has aroused interest in the area of electrical appliances and communication-related applications. However, due to their miniscule size, the excellent properties of these nanostructures can only be exploited if they are homogeneously embedded into lightweight matrices as those offered by a whole series of engineering polymers. 17,18 CNTs hold the promise of delivering exceptional mechanical properties and multifunctional characteristics. 19 -21 Ever-increasing interest in applying CNTs in many different fields has led to continued efforts to develop dispersion and functionalization techniques. To employ CNTs as effective reinforcement in polymer nanocomposites, proper dispersion and appropriate interfacial adhesion between the CNTs and polymer matrix have to be guaranteed. 22
To maximize the advantage of CNTs as effective reinforcement for high-strength polymer composites, the CNTs should not form aggregates and must be well dispersed to enhance the interfacial interaction with the matrix. Several processing methods available for fabricating CNTs/polymer composites based on either thermoplastic or thermosetting matrices have been described in past review articles. 23 They mainly include solution mixing, in situ polymerization, melt blending, and chemical modification processes.
The most common method for preparing CNTs/polymer composites has been to mix both components into a certain solvent and evaporate the latter to form a composite film. The general protocol for all solution processing methods includes the dispersion of CNTs powder in a liquid medium by vigorous stirring and/or sonication, mixing the CNTs dispersion with a polymer solution and controlled evaporation of the solvent with or without vacuum conditions. In general, the most efficient dispersion of tubes during the first step is achieved by bath or tip sonication. Regarding thermosetting epoxy matrices, an early example of solution-based composite formation was described. 24 The present work describes the synthesis of three wholly aromatic polyamide–hydrazides which contained different predetermined amounts of para- and meta-oriented phenylene moieties, while still retaining the 1:1 content of amide and hydrazide linking bonds. The polymers have been prepared by the well-known low-temperature solution polycondensation reaction of para-aminobenzhydrazide (PABH) or meta-aminobenzhydrazide (MABH) and an equimolar amount of either terephthaloyl chloride (TCl), isophthaloyl chloride (ICl), or their appropriate mixtures in N, N-dimethylacetamide (DMAc) as a solvent. The effect of incorporation of 0.1 wt% MWCNTs into the prepared polymers on the thermal and electronic transition properties of the resulting composites is studied. The morphology and distribution of MWCNTs within the composites are investigated by field-emission scanning electron microscopy (FE-SEM), transmission electron microscopy, and atomic force microscopy (AFM) observations.
Experimental
Reagents and solvents
PABH (98%) and MABH (97%) were purchased from Alfa Aesar, UK. Calcium hydride, TCl, and ICl were purchased from Acros, Germany. Phosphorous pentaoxide (97%) was purchased from Alpha Chemika, Mumbai, Maharashtra, India. Multiwalled carbon nanotubes (MWCNTs) 40–60 nm were supplied by Nano Amour, Los Alamos, New Mexico, USA. Methanol (extra pure) and ethanol (99.9%) were purchased from Specific Drilling Fluids (SDF), Gandhidham Gujarat, India. DMAc (99.9%) obtained from Acros, part of Thermo Fisher Scientific, NJ, USA, was guaranteed reagent, dried over calcium hydride for 24 h, and followed by distillation under reduced pressure. The fractions, which boiled at 40–42°C/2 mmHg, were collected and stored over molecular sieves before use.
Polymerization procedure
All polymers were prepared by the same experimental procedure, which will be given here for preparation of polymer I. A dry 250-ml four-necked round bottom flask equipped with a Teflon-coated half-moon mechanical stirring bar, a thermometer, and a drying tube was placed in a dry box. MABH, 3.02 g (0.02 mol), was charged into the reaction flask and followed by 80 ml of dry DMAc. Stirring was started until complete dissolution was achieved. The reaction flask was placed in a crushed ice-salt water bath and cooled at −10°C for 15 min. Then, 4.04 g (0.02 mol) solid ICl was added slowly under constant stirring over a period of 1 h by means of a funnel with weighing paper that was positioned just slightly above the surface of the stirred polycondensation reaction solution. After this period, stirring was continued for another 2 h at −10°C. Then the cooling bath was removed and the temperature of the polymerization reaction was allowed to rise gradually to room temperature and maintained for 24 h with constant stirring. Afterwards, a clear, slightly yellow viscous solution was obtained. Finally, the polymer solution was slowly poured into 600 ml methanol that was rapidly stirred. A fibrous white precipitate of polymer I immediately was formed. The polymer was isolated by vacuum filtration, washed successively with methanol, and dried in a vacuum oven at 75°C to constant weight. All the prepared polymer samples were purified by repeated precipitation from their solutions in DMAc using methanol as a nonsolvent. The preparation of the polymers and their structures is illustrated in equations 1–3.
Film preparation
As-prepared polymers films casting techniques
Films were prepared by casting 5% wt/vol polymer solutions in DMAc onto dry clean Pyrex glass plates to a uniform thickness. Solvent evaporation was performed at constant temperature of 100°C in an electrically heated oven with forced air circulation. The resulting films were kept in the oven until no change in weight could be observed. The films were then immersed in deionized water overnight to remove any residual solvent. Finally, the films were dried in a vacuum oven at 75°C to constant weight. The thickness of the films was 0.04 mm ± 0.01 mm, measured by a digital micrometer.
MWCNTs/polymer composite films preparation
MWCNTs/polymer nanocomposite films were prepared by mixing the polymer solutions in DMAc (5% wt/vol) with 0.1 wt % MWCNTs (based on the polymer weight). The produced polymer nanocomposite solutions were sonicated for 3 h and then stirred overnight. Then the polymer nanocomposite solutions were cast onto dry clean Pyrex glass plates to a uniform thickness. Solvent evaporation was performed at constant temperature of 100°C in an electrically heated oven with forced air circulation as previously stated. The resulting films were kept in the oven until no change in weight could be observed. The films were then immersed in deionized water overnight to remove any residual solvent. Finally, the films were dried in a vacuum oven at 75°C to constant weight. The thickness of the films was 0.04 mm ± 0.01 mm measured by a digital micrometer.
Polymers identification
Infrared spectra of the prepared polymers were measured on a Thermo Fourier transform infrared (FTIR) Nicolet avatar 370 CSI (Waltham, MA, USA). The samples were measured as pellets with moisture-free KBr. All spectra were recorded within the wavenumber range of 4000–600 cm−1 at 25°C. Proton nuclear magnetic resonance (1H-NMR) spectra of the prepared polymers have been measured on Mercury300 NMR using dimethyl sulfoxide (DMSO) (Online Supplemental File).
Elemental analyses of the prepared polymers were performed at the Micro Analytical Unit of the Faculty of Science, Cairo University.
Polymer characterization
Thermogravimetric analysis
Thermogravimetric analysis (TG) curves were recorded on a TA Thermogravimetric Analyzer (TGA) Q500 (New Castle, DE, USA) under nitrogen atmosphere with a flow rate of 40 ml min− 1, at a heating rate of 10°C min− 1, and a heating range from room temperature to 800°C. The samples were thin films and their weights ranged from 3 mg to 5 mg.
Electronic transition properties
The optical properties of polymer/MWCNTs composite films prepared by dissolution technique were studied in the visible and ultraviolent wavelength regions from 200 nm to 800 nm using double-beam spectrophotometer Lambda 35 Perkin Elmer (Waltham, MA, USA).
Field emission analysis
The surface morphology of the polymer/MWCNTs composite films was investigated using QUANTA 250 S FEG microscope (Hillsboro, OR, USA). Polymer thin films were coated with gold. Energy used in the microscope is 5–10 keV with magnifications ranged from 5000 to 50,000×.
Atomic force microscopy
AFM images of polymer I/MWCNT composites were recorded on AFM (scanning probe microscope (SPM), Shimadzu, Japan) using noncontact mode.
Results and discussion
This article deals with the preparation of three wholly aromatic poly (amide–hydrazides) and their nanocomposites with MWCNTs. Characterizations including the spectral, morphological, thermal, and optical properties of the resulting nanocomposites were discussed in terms of the structure relationships.
Polymer synthesis
Three wholly aromatic poly(amide–hydrazides) have been synthesized by a low-temperature (at −10°C) solution (in anhydrous DMAc) polycondensation reaction of either MABH or PABH with stoichiometric amount of ICl producing polymer I (100% meta-phenylene rings content) and polymer II (50%: 50% meta-: para-phenylene rings content), respectively. The third polymer, polymer III (75%: 25% para-: meta-phenylene rings content) was prepared by condensing PABH with mixture of equimolar ratio of TCl and ICl. The para- and meta-phenylene moieties content were altered within these polymers so that the changes in the latter were 50 mole% from polymer I to polymer II and were 25 mole% from polymer II to polymer III, starting from an overall content of 100–25 mole%. Thus, all the polymers are structurally similar except for the linking mode of phenylene groups along the polymer chain.
The basic reaction employed here is the Schotten–Baumann condensation of an aromatic acid chloride and an aminohydrazide, which is generally known to be fast and quantitative. 25,26 These properties indicate that when this polymerization is performed by gradual addition of solid acid chloride in a solution of aminobenzhydrazide monomer, the hydrazide groups of the latter react first, and the so-called wholly ordered polymer with alternating amide and hydrazide linkages is formed. The liberation of HCl by-product has multifunctional effects on this reaction. First, HCl acts as a reaction catalyst. Second, it reacts with the solvent (DMAc) to form salt (DMAc–HCl). 27 However, it could be expected that there is a competition between the growing polymer molecules and the solvent (DMAc) for HCl. The solvent is more basic than the polymeric product, thus associating with solvent carbonyl oxygen predominates. The forming salt is useful in enhancing the solubility of the growing polymer, thereby assisting in molecular weight buildup and should direct the polycondensation equilibrium toward completion and be an additional driving force for the process.
Consequently, since the basic reaction is rapid even at low temperatures and, since one of the polymerization products is continuously being withdrawn from the equilibrium, this condensation polymerization reaction is expected to proceed to acceptably high conversions and thus satisfy one of the mentioned requirements for preparation of high molecular weight products imposed by the nature of this polycondensation reaction.
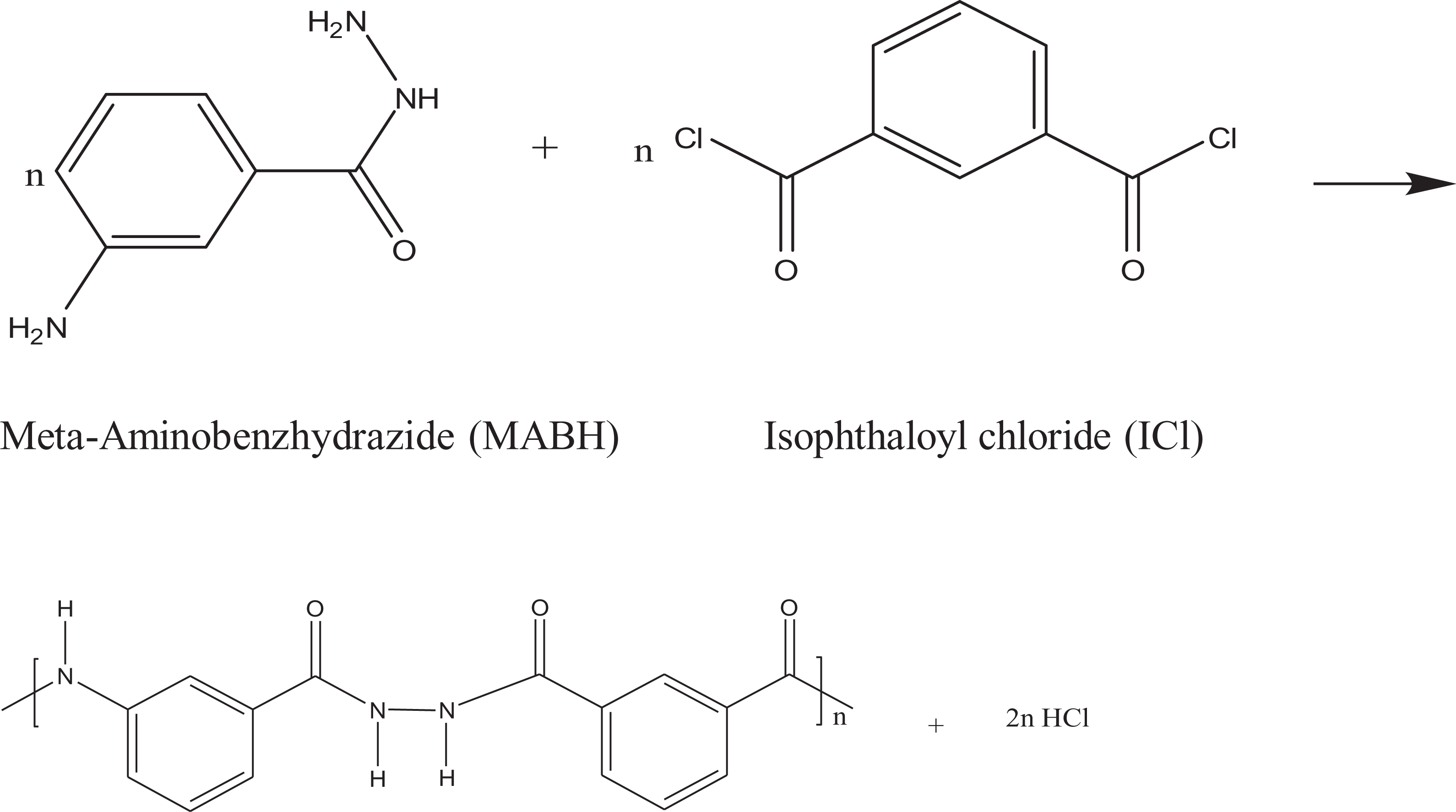 Eq.1
Eq.1
 Eq.2
Eq.2
 Eq.3
Eq.3
Polymer identification
FTIR analysis of the prepared polymers
The structure of the prepared polymers is verified on the basis of their FTIR spectra and elemental analyses. All the FTIR spectra of these polymers (Figure 1) showed common absorption stretching vibration bands at the following wavenumbers: 3430–3260 cm−1 (intensive and broad) assigned the overlapped =NH and the possible interchain hydrogen bonding; 2300–2180 cm−1 (weak) is attributed to possible enol-type configuration of hydrazide and amide groups; 1660–1640 cm−1 (strong) corresponded to the amide carbonyl group; 1580–1600 cm−1 indicated the aromatic carbon–carbon double bonds; 1525–1515 cm−1 is due to =NH of amide II; 1500 cm−1 indicated carbon–carbon single bond in ring, and 1340, 1280, and 1250 cm−1 corresponded to carbon–hydrogen combined with =NH of amide III. 28,29

FTIR spectra of the prepared wholly aromatic polyamide–hydrazides.
1H-NMR analysis of the prepared polymers
1 H NMR spectra measured in DMSO for the as-prepared polymers, polymer I, polymer II, and polymer III, respectively (Online Supplemental File). For amide and hydrazide protons, the chemical shifts are observed between 10 ppm and 11 ppm. This indicates a successful synthesis of as-prepared polymers. For aromatic hydrogen (ArH) chemical shift, polymer I shows a constitutional order of chemical shift between 7 ppm and 8 ppm, due to regularity of polymeric chain (completely meta-oriented polymer). Polymer II shows partial disorder of resonating chemical shift of ArH, due to incorporation of para-phenylene moiety by a 50% into the repeating unit structure. This was observed between 7 ppm and 8 ppm. Polymer III showed multiple resonance for the ArH between 7 ppm and 8 ppm. This could be attributed to irregularity of the polymeric chains (25% meta- and 75% para-aromatic moieties). Thus, nonequivalent ArHs. Also as-prepared polymers show a chemical shift at 3.2 ppm due to adsorbed water resulting from the hygroscopic structure of polymeric chains. Also, for peaks at 2.4 ppm, this could have attributed to rare traces of dimethyl acetamide used for polymers synthesis.
Elemental analysis of the prepared polymers
On the other hand, the elemental analysis values are in good agreement with the theoretical ones calculated for the expected repeating unit (C15H11N3O3) as presented in Table 1.
Elemental analysis of as-prepared aromatic polyamide hydrazides.

a Calculated as %O = 100−(%C + %H + %N).
Polymer characterization
Thermal stability
Normalized weight losses and differential thermogravimetric (DTG) curves measured in nitrogen atmosphere using TG analysis technique for the prepared polymers and their corresponding MWCNT composites are plotted in Figures 2(a) and (b) and 3(a) and (b). All polymers and their MWCNT composites showed a characteristic similar thermal behavior which consisted of three distinct stages through which appreciable weight losses were observed.

(a) TG curves of the prepared wholly aromatic polyamide–hydrazides in nitrogen at a heating rate of 10°C min−1. (b) DTG curves of the prepared wholly aromatic polyamide–hydrazides in nitrogen at a heating rate of 10°C min−1. TG: thermogravimetric; DTG: differential thermogravimetric.
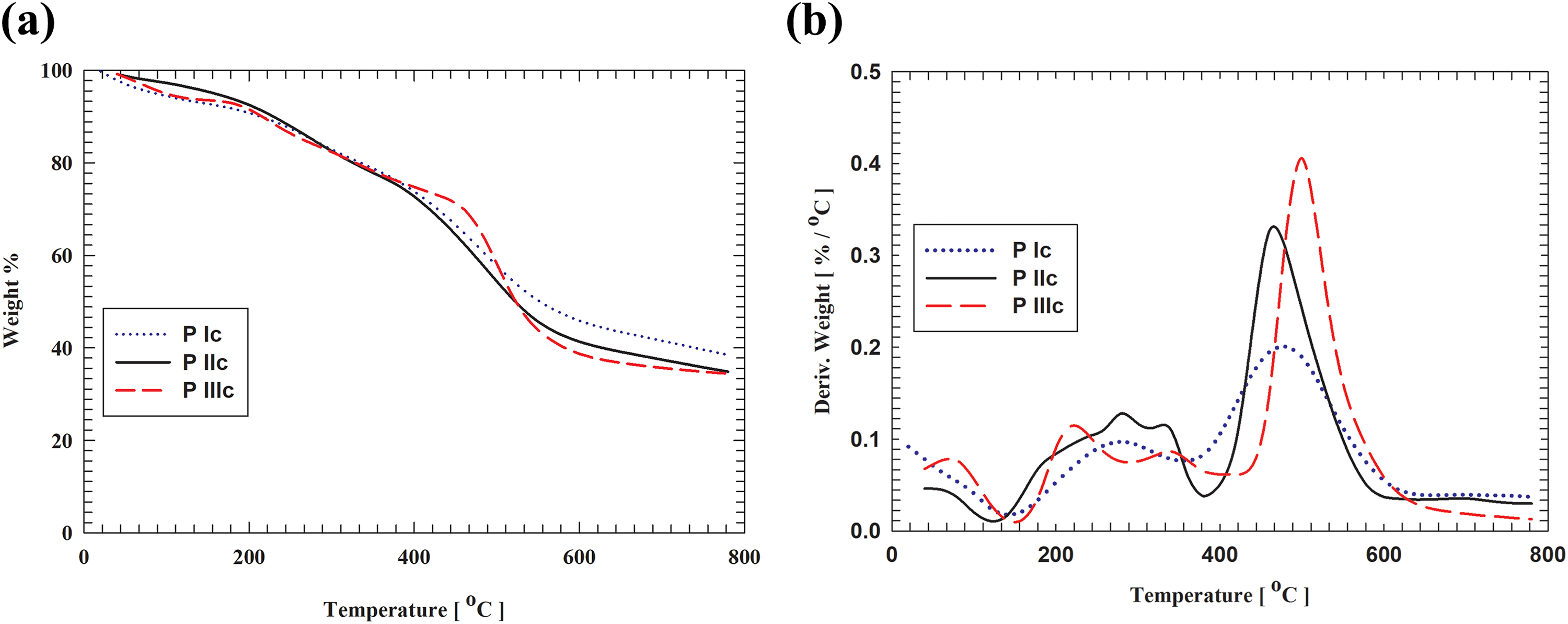
(a) TG curves of the polymers + 0.1 wt% MWCNTs in nitrogen at a heating rate of 10°C min−1. (b) DTG curves of the polymers + 0.1 wt% MWCNTs in nitrogen at a heating rate of 10°C min−1. TG: thermogravimetric; DTG: differential thermogravimetric; MWCNTs: multiwalled carbon nanotubes.
Thermal stability of the as-prepared polymers
TG and DTG data of the free polymers (Figure 2(a) and (b), respectively), showed firstly, weight loss of 5–6 wt% (based on the original polymer weight) occurred at 60–110°C and assigned to evaporation of adsorbed water from the surface of the polymer sample. Secondly, significant weight losses of 14–25 wt% for polymer I, 13–20 wt% for polymer II, and 12–20 wt% for polymer III (based on the weight of the polymer samples) were obtained at temperature ranges from 245°C to 370°C, from 290°C to 380°C, and from 295°C to 400°C for polymers I, II, and III, respectively. These weight losses can be attributed to cyclodehydration reaction of the polymers hydrazide groups into 1, 3, 4-oxadiazole rings as represented in Figure 4. 27 -33 Also the residue of the solvent that makes strong physical interaction with the polymeric chains may explain the deviation of the weight loss at this stage from theoretical value (6–8%). Third, a steep weight loss took place at temperatures above 485°C for polymer I, 475°C for polymer II, and 480°C for polymer III and indicated the decomposition of the polymer containing 1,3,4-oxadiazole rings which were formed in the second stage. From these data, the order of thermal stability was polymer III > polymer II > polymer I. Thus, we can refer these results to the substitution of para-phenylene moieties for meta-phenylene ones in the chains lead to improved polymer stability at high temperatures. Moreover, this should also be associated with the regularity of super molecular packing within the bulk of the investigated polymers. In this case, the collinear arrangement of the para-phenylene units should allow for establishment of stronger intermolecular hydrogen bonds which would be more difficult to break and, therefore, more resistant to elevated temperatures.32.33 On the other hand, for the highest initial decomposition temperature, we found that the order of thermal stability was polymer I > polymer III > polymer II and for mass residue percent at 800°C was polymer II > polymer I > polymer III. This may be due to the regularity of the chain structures that is due to the repeat units arrangement affect to great extent the thermal stability. In this case, the more possible formation of interchains hydrogen bonds in a given polymer the more stable the thermal stability. Hydrogen bonds between the chains should result in more restriction in the freedom of chain lateral movement upon heating. Spatial distribution of polymer chains of polymer I should allow this situation over the other two polymers in case of initial decomposition. In case of mass residue, and if we assume that at or above this temperature not only bonds disintegration takes place but also ring rupture is also possible. Therefore, apparently in case of polymer II, there are more restrictions in the chains to allow the final disintegration of the polymer.

Cyclodehydration and thermal degradation reaction of the prepared wholly aromatic polyamide–hydrazides.
Thermal stability of polymer composites
Normalized weight loss curves measured in nitrogen atmosphere using TGA technique for the MWCNTs composite samples were plotted in Figure 3(a) and (b). The data showed that the highest initial degradation temperature of polymer III/MWCNTs (IIIc) was 500°C, of polymer II/MWCNTs (IIc) was 465°C, and of polymer I/MWCNTs (Ic) is 480°C. The final mass residue of Ic was 38.5% and for IIc and IIIc was approximately 35%. From the above results, we can conclude that the incorporation of MWCNTs into IIIc matrix enhanced thermal degradation behavior. This could be attributed to a barrier labyrinth effect of the nanotubes that the diffusion of degradation products from the bulk of the polymer to phase is slowed down 34 or transformation that started with relatively restricted chain mobility might end by transition to the gas phase.
It is important at this stage to compare the thermal analysis data of the as-prepared polymers with those of the composite films.
Normalized weight loss curves measured in nitrogen atmosphere using TGA for the free polymers and MWCNTs composite samples were plotted in Figures 5 (a) and (b), 6 (a) and (b), and 7 (a) and (b). From these curves, we noted that polymer I and its MWCNTs composite (Ic) showed that the onset degradation temperature of polymer I is approximately 485°C while in I/MWCNTs composite (P Ic) was 480°C. On the other hand, the mass residue at 800°C was 38.5% for I/MWCNTs composite (P Ic) compared to 32% of polymer I which is in a good agreement as previously reported. 35
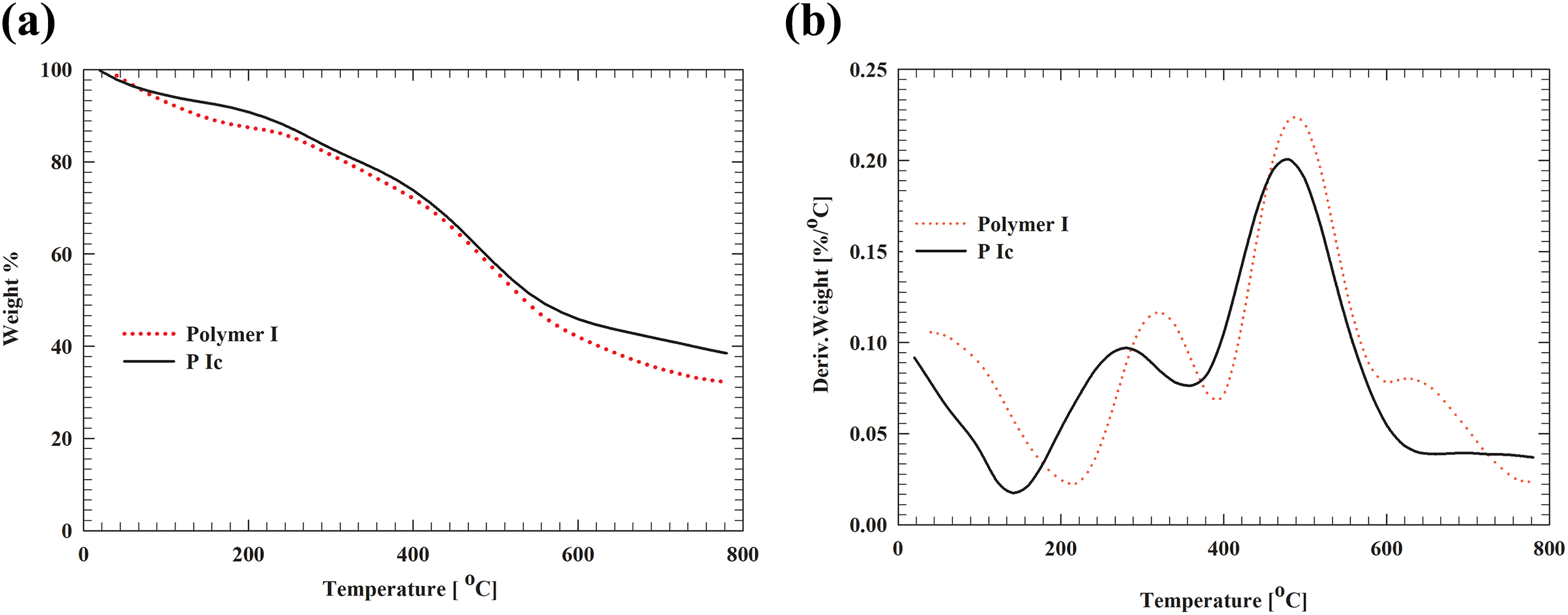
(a) TG curves of polymer I and polymer I + 0.1 wt% MWCNTs composite in nitrogen at a heating rate of 10°C min−1. (b) DTG curves of polymer I and polymer I + 0.1 wt% MWCNTs composite in nitrogen at a heating rate of 10°C min−1. TG: thermogravimetric; DTG: differential thermogravimetric; MWCNTs: multiwalled carbon nanotubes.

(a) TG curves of polymer II and polymer II + 0.1 wt% MWCNTs composite in nitrogen at a heating rate of 10°C min−1. (b) DTG curves of polymer II and polymer II + 0.1 wt% MWCNTs composite in nitrogen at a heating rate of 10°C min−1. TG: thermogravimetric; DTG: differential thermogravimetric; MWCNTs: multiwalled carbon nanotubes.

(a) TG curves of polymer III and polymer III + 0.1 wt% MWCNTs composite in nitrogen at a heating rate of 10°C min−1. (b) DTG curves of polymer III and polymer III + 0.1 wt% MWCNTs composite in nitrogen at a heating rate of 10°C min−1. TG: thermogravimetric; DTG: differential thermogravimetric; MWCNTs: multiwalled carbon nanotubes.
For polymer II and its MWCNT (P IIc) composite, the thermal behavior showed that the onset degradation temperature of polymer II is approximately 475°C, while II/ MWCNT (IIc) composite was 465°C. Also, mass residue at 800°C for II/MWCNTs composite (IIc) was approximately 35% compared to 34.5% of polymer II. Finally, for polymer III and its MWCNTs (P IIIc) composite, the thermal behavior showed that the onset degradation temperature of polymer III was approximately 480°C while in III/MWCNTs composite (P IIIc) was 500°C. The mass residue of III/MWCNTs composite (P IIIc) at 800°C was approximately 35% compared to 22% of polymer III which is also in good agreement as previously reported. 35 Also, this may be attributed to the fact that MWCNTs act as radical scavengers at low loadings that result in delaying the degradation pattern and enhancing the thermal stability. 36 It has also been reported that the enhancement in thermal conductivity on MWCNTs addition to the matrix that lowers the degradation temperatures. 37 The thermal stability of the MWCNT composites depends to a certain extent on the interaction between the phases. 38 For anomalous behavior of P Ic and P IIc composites, this may be due to sonication that may cause polymer chain fragmentation, formation of less stable γ-phase, or even reduction in CNT length. 39 We can also define T1 as the temperature at which the polymer loses 1% of its initial weight. Thus, as indicated in Table 2, the order of stability of the as-prepared polymers is I > II > III. Interestingly, the order is reversed when considering the polymers corresponding MWCNT composites. In this respect, the relative irregularity of the chains as represented by polymer III compared to polymers II and I showed less resistance to heating effect. While the incorporation of MWCNTs resulted in reversing the resistance of the corresponding composite that is due to the interfilling of the MWCNTs within the polymer chains. Therefore, the increase in thermal stability should be attributed to the development of more static forces between the chains.
Thermal stability of as-prepared polymers and their mwcnts composites after loss 1% of their initial weight.

MWCNTs: multiwalled carbon nanotubes.
Electronic transition properties of polymer composites
From Figures 8 (a) and (b) and 9 (a) and (b), we can state that for the prepared composite polymeric films, there are symmetrical ultraviolet–visible spectra. The cutoff absorbance bands were within the range between 370 nm and 350 nm (n−π*). The approximate λmax for cutoff band was 370 nm for P IIIc, 355 nm for P IIc, and 345 nm for P Ic. From visible region (400–800 nm), there is a slight difference in films transparency and their order of transparency is P Ic > P IIc > P IIIc. Compared to as-prepared polymers, there was a blue shift of values of λmax of cutoff band within a range of 10 nm due to inclusion of MWCNTs. This was explained previously as MWCNTs clusters affect in polymeric chain conjugation system due to SP2 carbon atoms of MWCNTs increased the energy of π−π* and n−π* of conjugated system through polymeric backbone, thus the λmax of cutoff band was shifted to lower wavelength (blue shift). 40

(a) UV-Vis spectra of the prepared wholly aromatic polyamide–hydrazides. (b) UV spectra of the prepared wholly aromatic polyamide–hydrazides. UV-Vis: ultraviolet–visible.

(a) UV-Vis spectra of polymer composites. (b) UV spectra of polymer composites. UV-Vis: ultraviolet–visible.
Surface morphology
SEM observations
Figure 10 at magnification 100,000× showed FE-SEM of polymer III + 0.1 wt% MWCNTs (P IIIc). The separation and dispersion of nanofillers were ascertained. However, as depicted in Figure 11 at magnification 5000×, FE-SEM image of polymer I + 0.1 wt% MWCNTs (P Ic) showed some clustered regions of filler appeared due to high energy exerted on the sample from the electron beam of the electron microscope. This resulted in some entanglement and random orientation of the filler. This creates interconnecting structures between neighboring strands. 18

FE-SEM image of polymer III + 0.1 wt% MWCNTs (P IIIc). FE-SEM: field emission scanning electron microscopy; MWCNTs: multiwalled carbon nanotubes.

FE-SEM image of polymer I + 0.1 wt% MWCNTs (P Ic). FE-SEM: field emission scanning electron microscopy; MWCNTs: multiwalled carbon nanotubes.

3-D image of AFM of polymer I + 0.1 wt% MWCNTs composite (P Ic). 3-D: three-dimensional; AFM: atomic force microscopy; MWCNTs: multiwalled carbon nanotubes.
AFM observations
The inclusion of the MWCNTs was also ascertained from AFM measurements. Three-dimensional pictures of polymer I + 0.1 wt% MWCNTs (P Ic) were given in Figure 12. The dispersion of the MWCNTs was again confirmed with good separation between the filler strands.
Conclusions
Three aromatic polyamide–hydrazides were synthesized via low-temperature polycondensation reaction, then 0.1 wt % MWCNTs was incorporated into these polymer matrices to enhance thermal and optical properties. FTIR data showed the presence of the =NH amide linkages within the polymer structure. NMR results ascertained the regularity of polymeric chains. From thermal analyses, the weight losses were due to cyclodehydration reaction of the hydrazide groups. Para-substitution within the polymer matrix resulted in enhanced thermal stabilities and the inclusion of MWCNTs increased the thermal characteristics of the composites. The optical transparency of the polymers should a decreasing order of I > II > III. The cutoff of the spectral pattern is in the 370–350 nm range. A blue shift was observed in the optical spectra upon the inclusion of MWCNTs to the polymer matrix.
Supplemental material
Supplemental Material, 1HNMR_SUPPLEMENTARY_FIGURE - Thermal and optical properties of aromatic polyamide–hydrazides modified with multiwalled carbon nanotubes
Supplemental Material, 1HNMR_SUPPLEMENTARY_FIGURE for Thermal and optical properties of aromatic polyamide–hydrazides modified with multiwalled carbon nanotubes by Nadia A Mohamed, SalahE Selim and Ahmed Galal in Polymers and Polymer Composites
Footnotes
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.