
Abstract
This study was performed to determine and compare the effect of heat and gamma-ray polymerization methods on the residual monomer and flexural strength of polyethylene fiber-reinforced denture-base acrylic resins. Four groups (n = 10) of specimens of polyethylene fiber-reinforced denture-base material were prepared in the form of thin disks. The first group was subjected to heat-curing and the other three groups were polymerized with gamma irradiation at doses of 15, 25, and 35 kGy, respectively. Fourier-transform infrared (FTIR) spectrometer was used to monitor the corresponding polymerization processes. The analysis of residual monomer was carried out by high-performance liquid chromatography–photodiode array detector. A three-point bending test was used to evaluate the flexural strength of the samples. The one-way analysis of variance test was performed to determine the significant differences between the groups. The absence of the bands related to carbon–carbon double bond in the FTIR spectra of all test groups was an evidence of polymerization. The mean weight percentage of residual monomer was successively ranked from highest to lowest in; heat-cured, gamma-cured at 15–35 kGy. However, no significant difference (p = 0.462) was found between gamma-cured samples at 25 and 35 kGy. Mechanical test results revealed that heat-cured samples had higher flexural strength than gamma-cured specimens (p < 0.001). Within the limitations of this study, polymerization with gamma-rays at 15 kGy was proposed as a promising technique in terms of the residual monomer and flexural strength results.
Keywords
Introduction
Poly(methyl methacrylate) (PMMA) (Figure 1) was first used as a denture base material in 1937. It has an extended history in dentistry due to its ease of handling, processing and polishing characteristics, acceptable aesthetics, low cost, and biocompatibility in an oral environment. 1 This denture base material is a multiphase system where methyl methacrylate (MMA) (Figure 1) monomer is mixed with prepolymerized powder beads of PMMA during fabrication. However, polymerization reaction ends up to a certain degree of monomer conversion, and therefore some unreacted monomers, called residual monomers, are left in the denture base polymer. Residual monomer left in the polymer may leach into saliva and cause adverse reactions in the oral mucosa adjacent to the denture. 2 Hence, many analyses using Fourier-transform infrared (FTIR) spectrometer, 3 -5 gas chromatography (GC), 6 -9 high-performance liquid chromatography (HPLC), 4,10 -15 and UV–Vis spectrometer 16,17 have been carried out to determine the degree of conversion and the levels of residual MMA monomer.
Residual MMA concentration varies depending on the methods and the conditions of polymerization. Chaves et al. 18 reported that heat-polymerized resins showed lower cytotoxic effects than autopolymerizing denture base acrylic resins and light-polymerized or dual-polymerized reline resins. According to Sheridan et al. 19 among the cytotoxic effects of chemically activated, heat-activated, and microwave-activated acrylic resins on gingival fibroblasts, the greatest cytotoxic effect was produced by chemically activated acrylic resins. Bayraktar et al. 20 revealed that autopolymerized resins eluted considerably more substances compared to the heat- and microwave-polymerized resins.
Chemical structure of MMA and PMMA.
Another problem of complete or partial denture prosthesis made from PMMA is most likely in the form of fracture either due to fatigue or impact forces of mastication. Therefore, several attempts have been made to improve the flexural and impact strength of PMMA. Research studies in this area have aimed at modifying the composition or reinforcing the PMMA with stronger materials such as metallic fillers, fibers, and developing new materials with better properties. 21 However, some studies have pointed out an increase of the residual monomer in reinforced polymers. 22 -25 Miettinen and Vallittu 25 reported that the release of residual MMA from heat-cured test specimens with glass fiber reinforcement was significantly higher than from unreinforced test specimens. It is difficult to predict the individual tolerance level of residual monomers for each person. Therefore, reinforced dentures that have relatively high residual monomer content compared with the unreinforced ones may prone to failure during clinical service. In this regard, the application of ionizing radiation as a polymerization method at an optimum dose may be a promising solution to this problem.
Ionizing radiation has long been used in polymer chemistry as a means of initiating polymerization, cross-linking, and decomposing particular polymer components. 26 Radiation-initiated polymerization has some advantages compared with conventional polymerization. These include the possibility of polymerizing any compound (monomer) that can be converted to polymer, the high purity of the product (since sensitizers and catalysts are absent), independence of initiation rate on temperature (radiolysis yields are only slightly temperature dependent), and ease of control of the reaction (e.g. by control of dose rate). 27
Gamma radiation-induced polymerization of PMMA is not a new phenomenon. 28 -30 Usanmaz et al. 31 have for the first time used the gamma radiation for curing of PMMA as a denture-base material and investigated the effect of the radiation dose on the thermal and rheological properties of the resin, however, residual monomer content in the samples has not been determined. Cehreli et al. 32 have investigated the cytotoxicity of eluates from gamma-ray-polymerized PMMA. They have reported that the eluates from PMMA polymerized by low doses of gamma-ray periods have early inhibitory effects on cell response, higher doses of gamma-irradiation lead to better cellular response, and therefore, may be future candidates for polymerization of PMMA. 32 However, further studies have only focused on the effect of ionizing radiation on the mechanical properties and color stability of different experimental denture base polymers. 33 -35 Consequently, there is still a lack of published information about the effect of gamma-ray polymerization on the residual monomer content in fiber-reinforced dental-base materials.
The aim of the present study was to determine and compare the effect of heat and gamma-ray polymerization methods on the residual monomer of polyethylene fiber-reinforced acrylic resins. Specimens were examined using FTIR and high-performance liquid chromatography–photodiode array detector (HPLC-PDA) system. In addition, the effect of gamma-ray polymerization on the flexural strength of polyethylene fiber-reinforced acrylic resins was also investigated and the relationship between residual monomer content and mechanical properties of the base was discussed.
Materials and methods
Materials
Heat-cure denture base materials were supplied by VertexTM Rapid Simplified (Vertex-Dental. BV, Zeist, the Netherlands) in the form of powder and liquid. HPLC grade methanol was purchased from Sigma-Aldrich (St. Louis, MO, USA) (CHROMASOLV®, for HPLC, ≥ 99.9%). High-purity water was prepared with a Milli-Q Gradient water-purification system (Millipore, Billerica, MA, USA).
Preparation of standards and samples
Four groups (n = 10) of specimens of denture-base material were prepared in the form of thin disks of dimensions 60 × 10 × 4.0 mm3 as recommended by the manufacturer. Cold gas plasma-treated woven ultra-high modulus polyethylene fibers (Ribbond, Ribbond Inc., Seattle, Washington, USA) were first wetted with MMA liquid and then placed in the middle of the acrylic doughs. The first group was subjected to heat-curing and labeled with “HC.” Other three groups were polymerized with gamma irradiation at doses of 15, 25, and 35 kGy and labeled with “GC15,” “GC25,” and “GC35,” respectively.
Each specimen disk was broken into small pieces with an automatic grinding and weighed accurately with a balance accurate to five decimal places. Samples of approximately 650 mg were placed into separate one-mark 10 ml volumetric glass flasks. Samples were dissolved in acetone and agitated by magnetic stirring for 72 h at room temperature (23 ± 2°C) with a clean polytetrafluoroethylene (PTFE)-coated magnetic stirrer. After this process, to precipitate the dissolved polymer, methanol solution was added to each sample solution to a total volume of 10 ml. Solutions were filtered through a 0.45 µm filter and transferred into HPLC vials.
A stock solution of MMA was prepared by accurately weighing approximately 0.5 g monomer and dissolving the compound in methanol to give a final concentration of 5008.0 µg ml−1. Two sets of calibration solutions were prepared by diluting the stock solution to measure the low (in the range of 100.16–751.20 µg ml−1) and the high (in the range of 1201.08–2003.20 µg ml−1) residual MMA concentration.
Methods
The gamma irradiation was carried out in a Co-60 laboratory irradiator (Ob-Servo Sanguis, Institute of Isotopes, Budapest, Hungary), at doses of 15, 25, and 35 kGy (dose rate: 1.95 kGy h−1).
FTIR measurements in the range 4000–600 cm−1 were carried out using a Nicolet 8700 FTIR spectrometer (Thermo Scientific Instrument Co., Boston, USA) with an attenuated total reflectance accessory. The FTIR spectra were recorded with 32 scans at a resolution of 4 cm−1.
HPLC (Waters Alliance 2695 separations module; Waters, Milford, Massachusetts, USA) with PDA (Waters 2996 detector; Waters, Milford, Massachusetts, USA) were used to determine the amount of residual MMA content in the heat-cured and gamma-cured samples. Chromatographic separation was carried out on a C18 column (Waters Milford, MA, USA, XTerra® MS) (5 µm, 150 × 4.6 mm2) by employing an isocratic mobile phase comprising methanol/purified water (70/30, v/v). Before HPLC analysis, all solvents were degassed by ultrasonic treatment (Bandelin Sonorex, Berlin, Germany). The analytes were detected at a flow rate of 1.0 ml min−1. The injection volume of the samples was 10 μl. Column and detector were kept at 25ºC.
In the sample chromatogram, the peak having the same retention time as the standard MMA accepted as residual MMA. Linear regression equations obtained from calibration graphs of low and high standard solution sets were used to determine the concentration of MMA in micrograms, CMMA, per milliliter of analyzed sample solutions. The weight of the residual MMA in the sample was found from the dilution and unit conversion factor calculations. The weight percentage of the residual MMA, wt%, was calculated by proportioning the weights of residual MMA and sample.
The flexural strength tests were performed with a universal testing machine (Lloyd LRX, Lloyd Instruments, Fareham Hants, UK) using a three-point bend test. A crosshead speed of 5 mm min−1 was utilized and the distance between the specimen supports was 60 mm. Load was applied at the midpoint of the specimens and fracture force was recorded in Newtons (N). The flexural strength of each specimen was calculated (MPa) using the formula:
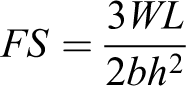
where FS is the flexural strength (MPa), W is the maximum load before fracture (N), L is the distance of the two supports (mm), b is the width of the specimen (mm), and h is the height of the specimen (mm).
Means and standard deviations were calculated for each group. To determine whether the differences between each processing technique were statistically significant, the one-way analysis of variance (ANOVA) (α = 0.05) test was conducted. A p value less than 0.05 was considered statistically significant.
Results
Fourier-transform infrared
FTIR has been used to monitor the corresponding polymerization processes. Figure 2 shows the FTIR spectra of the heat and gamma-ray polymerized groups. The main bands observed for all groups have been identified as follows: The band at 2995cm−1 is attributed to asymmetric C–H stretching of the methyl groups. The band observed at 2950 cm−1 is composed of symmetric stretching of methyl and asymmetric stretching of methylene overlapping bands. The weak absorption band at 2848 cm−1 is assigned to symmetric C–H stretching vibration of sp3 hybridized backbone methylene group (–CH2–). The carboxyl vibrations in the pendant group are characterized by the intense band at 1724 cm−1. The methyl and methylene bending vibrations are clearly observed at 1500–1300 cm−1 region, while the rocking peaks of these groups are located between 985 and 840 cm−1. The band at 750 cm−1 is attributed to the long-chain methyl [(CH2) n ] bending (rocking), which is also indicative of polymerization. From Figure 2, it can be seen that there are prominent absorption bands centered at 1238 and 1141 cm−1 and shoulders at 1267, 1190, and 1062 cm−1. All these bands are assigned to the C–O–C asymmetric and symmetric stretching vibrations.

FTIR spectra of the heat and gamma-ray polymerized samples.
The stretching vibration of the C=C bond usually gives rise to a moderate band in the region 1680–1600 cm−1. The C–H stretching frequency of a sp2 hybridized C–H (C=CH2) is at 3000–3250 cm−1 (higher than for sp3 hybridized C–H). In addition, a sp2 hybridized C–H (C=CH2) is also characterized by a strong band at approximately 900 cm−1. The absence of these bands in the spectra is an evidence of polymerization. Moreover, all sample groups have a residual monomer content below the detection limit of FTIR.
Apart from these bands, an additional band at 2916 cm−1 has been observed in the FTIR spectrum of GC35. Moreover, it can be clearly seen in the same spectrum that the band at 2848 cm−1 is significantly more intense than the corresponding band in the spectra of other samples. These spectral differences indicate the formation of structural changes in the sample irradiated at a dose of 35 kGy. In the FTIR spectrum, CH2 symmetric and asymmetric stretching vibrations give rise to the bands centered near 2850 and 2920 cm−1, respectively. 36,37 Therefore, the new band at 2916 cm−1 and relatively intense band at 2848 cm−1 are attributed to CH2 stretching vibrations and an observed increase in the intensity of these bands indicates the formation of additional CH2 groups (discussed in the fourth section).
High-performance liquid chromatography
Two calibration curves, prepared from two series of solutions with known concentrations of MMA monomers, were used to predict any unknown MMA monomer concentrations from the area of MMA peaks in the chromatograms. The calibration curves for low and high concentration range are shown in Figure 3(a) and (b). The equation and R2 value of calibration curves for low and high concentration range are y = 18,414x − 574,942, R2 = 0.9994 and y = 8861.8x + 7 × 106, R2 = 0.9993, respectively.

Calibration curves for (a) low concentration range, (b) high concentration range, and (c) combined calibration graph.
The reason for plotting two calibration curves is to get the best fit linear lines and to obtain R2 values close to 1 for both low and high standard solution sets. Since the calibration sensitivity (slope of the calibration curve) of high concentration set is lower than that of low concentration set, the combined calibration graph (Figure 3(c)) does not give a straight line over the whole concentration range. An R2 value of this graph is very low (0.9764), which reduces the accuracy of the results.
Figure 4 shows the representative chromatograms of heat-cured and gamma-cured samples. The peak areas of the residual monomer obtained from the thermally polymerized samples fit the calibration graph of the high concentration range, whereas the area of the residual MMA peak of gamma-cured samples lie in the calibration curve plotted for low concentration range. Therefore, for an accurate concentration determination, the equation of calibration curve “a” and “b” were used for the gamma-polymerized and heat-cured samples, respectively. The weight percentages of the residual MMA, wt%, were calculated and summarized in Table 1.
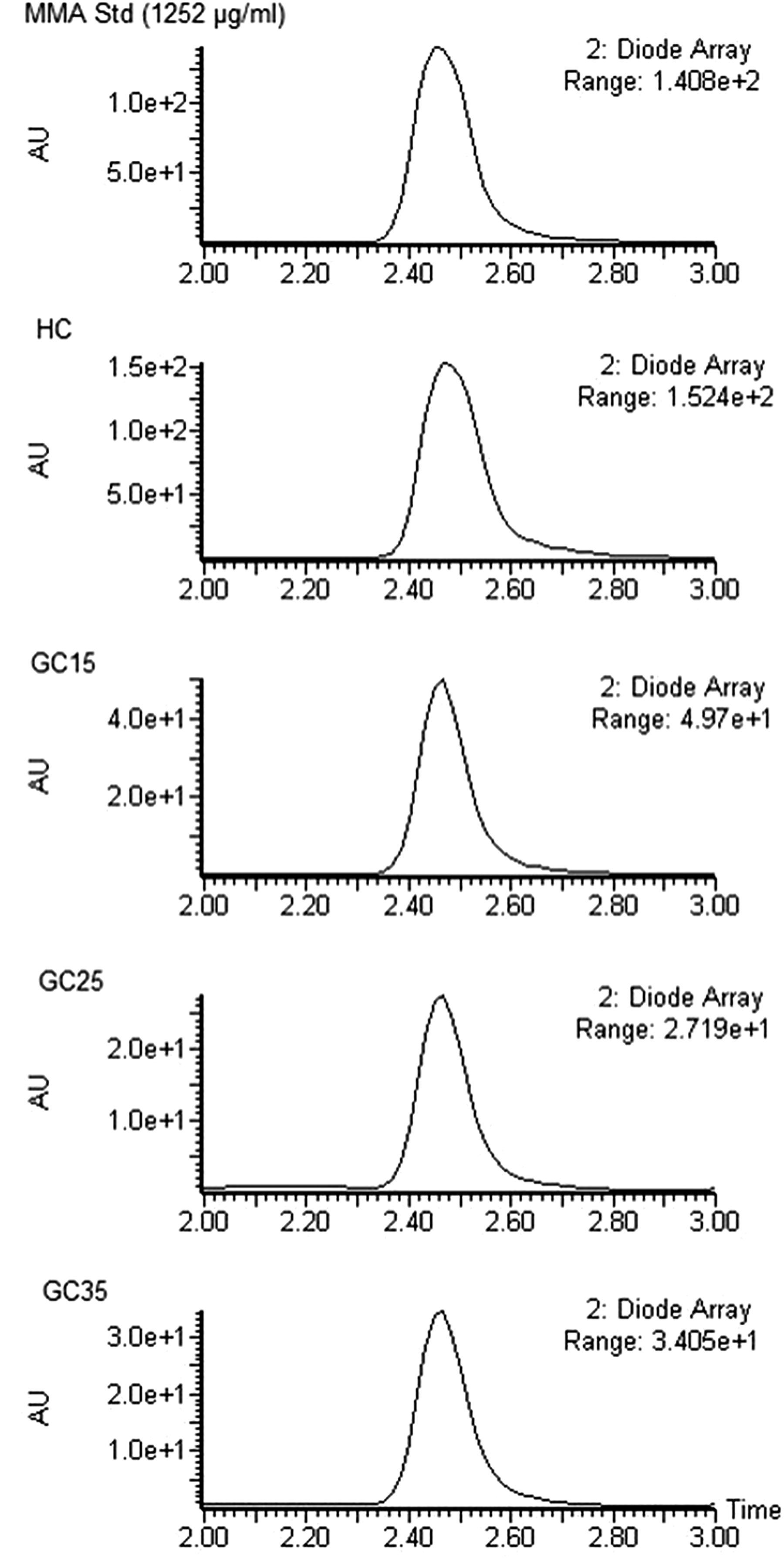
Representative chromatograms of MMA standard solution, heat-cured and gamma-cured samples.
The residual MMA (wt%) of heat-cured and gamma-cured sample groups expressed as mean values with standard deviations.

MMA: methyl methacrylate; HC: heat-cured; GC: gamma-cured (15, 25, and 35 kGy); MV: mean value; SD: standard deviation.
Mean values of the weight percentages of the residual MMA in HC, GC15, GC25, and GC35 samples were found to be 3.68, 0.94, 0.67, and 0.64, respectively (Table 1). The results of the one-way ANOVA of residual MMA are given in Table 2. The weight percentage of the residual monomer in HC samples is significantly higher than in GC15 (p < 0.001), GC25 (p < 0.001), and GC35 (p < 0.001) samples. According to the statistical results of residual monomer content of gamma-cured samples, residual MMA at 15 kGy is significantly higher than monomer content at 25 kGy (p < 0.001) and 35 kGy (p < 0.001). There is no significant difference between gamma-cured samples at 25 and 35 kGy (p = 0.462) (Table 2).
One-way ANOVA results of residual MMA for all groups.
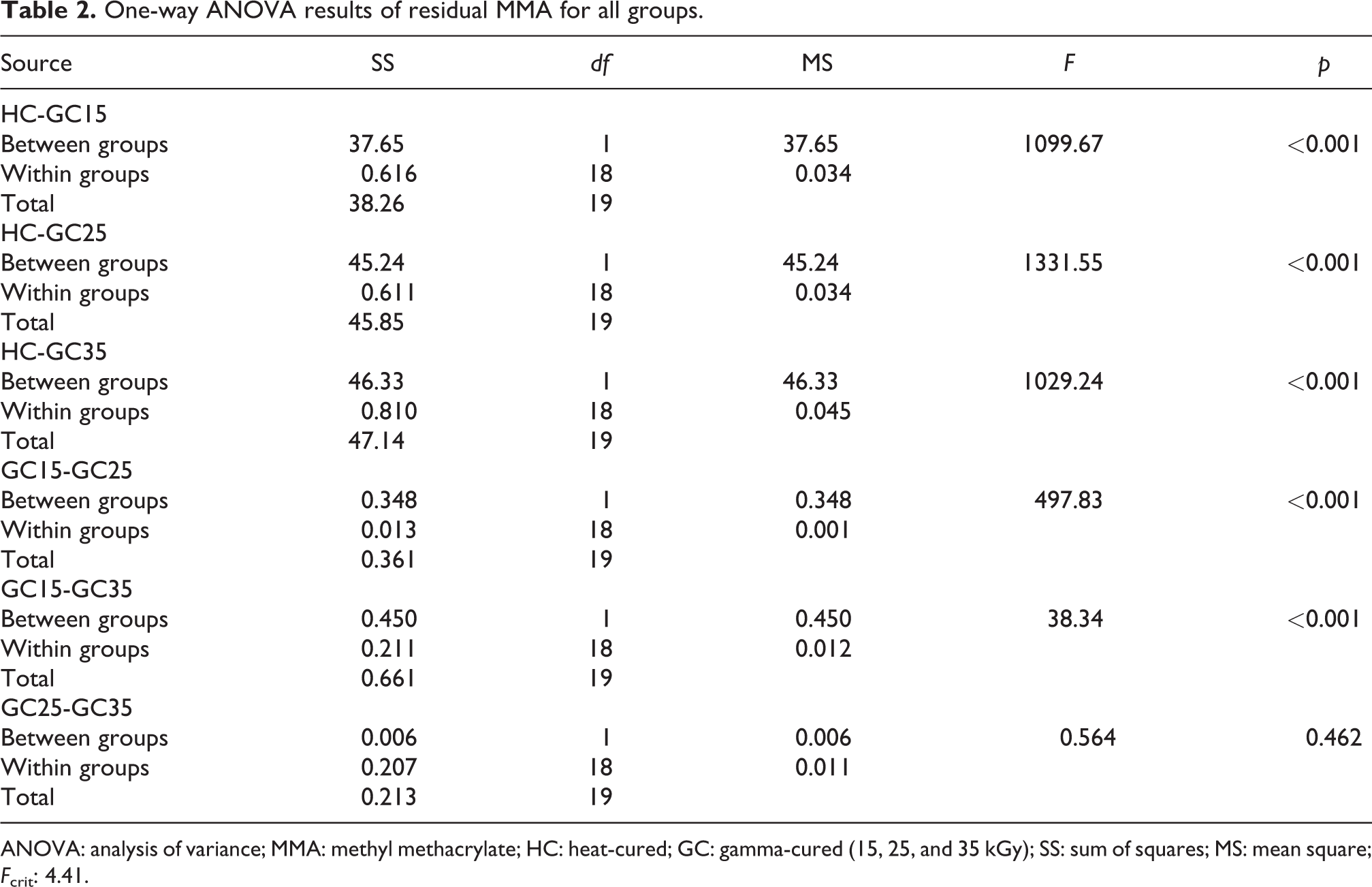
ANOVA: analysis of variance; MMA: methyl methacrylate; HC: heat-cured; GC: gamma-cured (15, 25, and 35 kGy); SS: sum of squares; MS: mean square; Fcrit: 4.41.
Table 3 shows the mean and standard deviation of the flexural strength values (in MPa) in the heat- and gamma-cured samples. HC group had the highest flexural strength and the GC35 group had the lowest flexural strength. The one-way ANOVA results are presented in Table 4. HC samples showed a significantly higher flexural strength (94.58 MPa) compared to the GC15 (77.90 MPa) (p = 0.018), GC25 (38.83 MPa) (p < 0.001), and GC35 (24.50 MPa) (p < 0.001). GC35 group exhibited the lowest flexural strength (p < 0.001). The one-way ANOVA indicated significant differences among the gamma-cured denture base materials in flexural strength (Table 4); GC15 exhibited higher flexural strength value than GC25 (p < 0.001) and GC35 (p < 0.001). GC25 showed significantly higher flexural strength compared to GC35 (p < 0.001).
The flexural strength (MPa) of heat-cured and gamma-cured sample groups expressed as mean values with standard deviations.

HC: heat-cured; GC: gamma-cured (15, 25, and 35 kGy); MV: mean value; SD: standard deviation.
One-way ANOVA results of flexural strength for all groups.

ANOVA: analysis of variance; HC: heat-cured; GC: gamma-cured (15, 25, and 35 kGy); SS: sum of squares; MS: mean square; Fcrit: 4.41.
Discussion
As mentioned in the section “Fourier-transform infrared,” lack of the peaks at 3000–3250 and 1680–1600 cm−1 region in FTIR spectra of heat- and gamma-cured (at all applied doses) samples indicates that the polymerization is substantially complete. On the other hand, the additional peaks observed in the spectrum of GC35 are evidence of the structural modifications that occurred at 35 kGy. These structural changes would seem to suggest the cross-linking of the polymer structure.
In the heat-curing process, the initiator is activated by heating the water to a very high temperature, which leads the polymerization reaction. Polymerization using gamma-rays also proceeds via the same mechanism as the free radical initiation mechanism. The energy absorbed from ionizing radiation gives rise to the formation of monomer radicals. The new monomer free radical reacts with another monomer molecule forming the chain propagation step to increase the length of the polymer chain. However, besides the polymerization, creation of new intra-chains chemical bonds (cross-linking), breaking of the chemical bonded structure (degradation), changes in unsaturation (the formation of double bond between carbon atoms), and oxidation (in the presence of air or oxygen) processes may take place during the irradiation. 38 These reactions are in competition and the predominance of these reactions depends on the absorbed doses and dose rate. As can be seen from the FTIR spectra of HC, GC15, and GC25 samples, the typical stretching and bending absorptions have remained relatively unchanged. It can be suggested that the prevalent reaction is polymerization in these samples and another type of process has not significantly occurred or is no FTIR-detectable. On the other hand, the FTIR spectrum of GC35 points out that cross-linking has become prominent at 35 kGy. The increase in intensities of asymmetric and symmetric stretching vibrations of CH2 (at 2916 and 2848 cm−1, respectively) could be explained by the cross-linking mechanism, leading to the formation of new intermolecular –CH2–CH2– bonds. 39-40 Plaček and Szőcs 41 used the electron spin resonance (ESR) method for studying radicals generated in PMMA by gamma-irradiation and they observed several types of radicals. In our opinion, one of the main radical they detected, –COOCH2·, is probably responsible for the cross-linking observed in this study. Therefore, it is suggested that the cross-linking reaction mechanism has proceeded through radiation-induced hydrogen abstraction from the methyl groups and coupling of two methyl radicals in adjacent PMMA molecules.
Apart from this mechanism, irradiation can result in chain scission of the main chain to give two end macro radicals (primary and tertiary radicals). These radicals may give unsaturated compounds (–C=C–) by intramolecular hydrogen abstraction 42 or oxygenated products (alcohols, aldehydes, and ketones) by addition of oxygen to these radicals. 43 However, the lack of identifiable bands related to carbon–carbon double bonds, suggested as an evidence of polymerization, also indicates that unsaturated compounds have not significantly formed. In addition, the absence of a very broad and strong O–H stretching band in the region 3500–2500 cm−1 corresponding to alcohols as well as no significant change in the intensity and position of the band at 1724 cm−1 corresponding to carbonyl compounds such as aldehydes and ketones, moreover, the absence of a doublet C–H stretching frequency band of aldehydes appears at 2750 and 2850 cm−1 are the evidence for the non-existence of oxygenated products. The absence of any effect of oxygen on PMMA degradation is due to the dose rate of the radiation source (1.95 kGy h−1). At this dose rate, diffusion of oxygen into PMMA is probably too slow to be a factor.
According to FTIR results, as a consequence, it is suggested that polymerization is the main reaction mechanism during the heat- and gamma-curing processes, however, cross-linking become prominent in gamma-cured samples at 35 kGy.
In the previous studies, 8,12,13,15,17,44 the amount of residual MMA in heat-cured samples was found generally less than the 2.2 mg% as required by ISO20795-1:2013. However, in the present study, the weight percentage of residual MMA in heat-cured reinforced resins varied between 3.96% and 3.07% (Table 1), which were considerably higher than the accepted average. Our findings are in agreement with the result of Miettinen and Vallittu 25 which points to the increase of the residual monomer content in reinforced resins. The addition of untreated fibers to the resin matrix can result in void formation between the resin and the fiber interface due to improper wetting of the fiber by the resin. As stated by Miettinen and Vallittu, 25 this problem has solved by decreasing the viscosity of the PMMA-MMA mixture used to wet the fiber, however, this causes an increase in the amount of residual MMA of the fiber-PMMA composite. Although this wetting process has been applied in this study, low amounts of residual monomer have been detected in gamma-cured samples due to the high penetration depth of the radiation, indicating polymerization with gamma-rays is a very effective technique for decreasing the monomer residue in denture base resins. At this point, our findings also highlight the significance of the dose value that affects the properties of the final product. The mean weight percentage of residual monomer is successively ranked from highest to lowest in; HC, GC15, and GC25. However, no significant difference (p = 0.462) has been found between gamma-cured samples at 25 kGy and 35 kGy. Although the mean values of GC25 and GC35 are not statistically significant, a large variation in the data set of GC35 has been observed as a result of random cross-linking (Table 1). Cross-linking reactions cannot occur uniformly, particularly in bulk polymerization, because large molecules of reacted matter cannot diffuse rapidly. Sooner or later, molecules become much less mobile and can only react with their neighbors. Thus, an unreacted end might remain unreacted when opportunities around it are depleted or it reacts with the cross-link junction where its other end is attached, so a loop forms. In this way, the network can gain defects and these localized defects provide a substantial number of places in the network where a penetrant molecule might more easily rest or pass through. 45 In the present study, the network of GC35 samples probably has more defects than the other groups due to the prevalence of cross-linking reactions. The monomer molecules can easily penetrate into these places, therefore, some samples in the group have more/less monomer content than others, leading to a large variation in the data set of GC35. On the other hand, the weight percentage of residual MMA in GC15 and GC25 samples are less variable than in GC35 samples (Table 1). It is suggested that –COOCH2· radicals have formed with much lower yields at doses of 15 and 25 kGy, therefore, cross-linking has not become the primary reaction.
Among the desirable properties of denture material is a possession of an adequate flexural strength, which indicates the resistance to deformation or fracture of the bulk material under a flexural load. 46 According to ISO 1567:1999 standard, 47 flexural strength of denture-base polymers shall not be less than 65 MPa. In the current investigation, HC and GC15 samples meet the requirement of the standard. However, flexural strength values of GC25 and GC35 are less than 65 MPa. It has been observed a significant reduction in the flexural strength with the decrease in the residual monomer content of the samples. The level of the residual monomer in the resin, polymerization shrinkage, voids, and crack development could be the potential factors of the decrease in flexural strength. The presence of residual monomer could affect the flexural strength because of its plasticizing effect, 48 which induce some flexibility of the composite matrix. It is known that the amount of the plasticizer is critical, high levels have an inverse effect on the mechanical properties. It has been reported that the decrease of plasticizer adsorbed on the PMMA beads surface may cause increases in mechanical properties. 49 However, a plasticizer is required to overcome the brittleness of the composites. The lower flexural strength properties of gamma-cured resins compared to heat-cured samples may be due to their very low levels of residual monomer, which means loss of plasticizer and makes them more brittle. Although the residual monomer content of GC25 and GC35 is not statistically significant, the lower flexural strength of GC35 samples could be attributed to their partially cross-linked structures. It is known that the formation of new bonds between chains increases the effective molecular weight, further tangles and traps molecules, and may reduce the effective concentration of plasticizers. 50 As a result, in our opinion, GC35 group has shown the lower flexural strength due to the non-uniformly distribution of the residual monomer in the network.
Another possible reason for the lower flexural strength properties of gamma-polymerized samples compared to the heat-cured group may be the void formation. Since gamma-cured samples could not be kept under pressure during the polymerization process unlike the heat-cured samples, internal voids could be resulted. On the other hand, a better mechanical interaction between fibers and HC groups could be explained by the applied pressure, high temperature during heat polymerization, and lower polymerization shrinkage. 51
It should be noted that different curing methods affect polymerization shrinkage, which can be an important factor for void formation. In the process of polymerization, the adsorbed monomer liquid inside the fiber shrinks more than the acrylic resin surrounding it which causes separation between a composite resin and the fiber. In gamma-cured samples, this process may be more pronounced due to the high penetration of the gamma-rays that increases the polymerization in the fiber. Therefore, the fiber and the matrix interaction could be restricted and the load could not be transferred from the matrix to the fiber, resulted in the fracture of the specimen and led to less flexural strength value than in heat-cured samples. Among the gamma-polymerized samples, the decrease in the flexural strength value with the increasing dose could be explained by the fact that void formation may be more prominent with the increasing dose.
Conclusion
In this study, the amount of residual monomer in heat- and gamma-cured polyethylene fiber-reinforced PMMA has been investigated. The experimental results confirm that fiber reinforcement increases the residual monomer content in heat-cured resins. However, polymerization with gamma-rays has been proposed as a promising technique to solve this problem. Our research has underlined the importance of the applied dose and dose rate during the irradiation process. Although, the amount of residual monomer in all gamma-cured sample groups (GC15, GC25, and GC35) has found less than in heat polymerized specimens, it must be noted that the level of residual monomer in GC35 has varied within a wide range. FTIR results explain this inhomogeneous distribution with a cross-linking phenomenon, which has become prevalent at 35 kGy. In addition, irradiation at a relatively high dose rate has prevented the formation of oxygenated by-products. As a consequence, it has been suggested that irradiation with doses below 25 kGy is more convenient in terms of residual monomer. However, mechanical tests have shown that the flexural strength of GC25 and GC35 have failed to meet the minimum requirements of ISO 1567:1999 standard. Considering all of the results, it can be concluded that gamma-ray curing at 15 kGy can be used as an alternative method of curing fiber-reinforced denture-base acrylic resins. However, further studies will need to be performed in order to show the clinical applicability of the gamma-curing method.
Footnotes
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.