
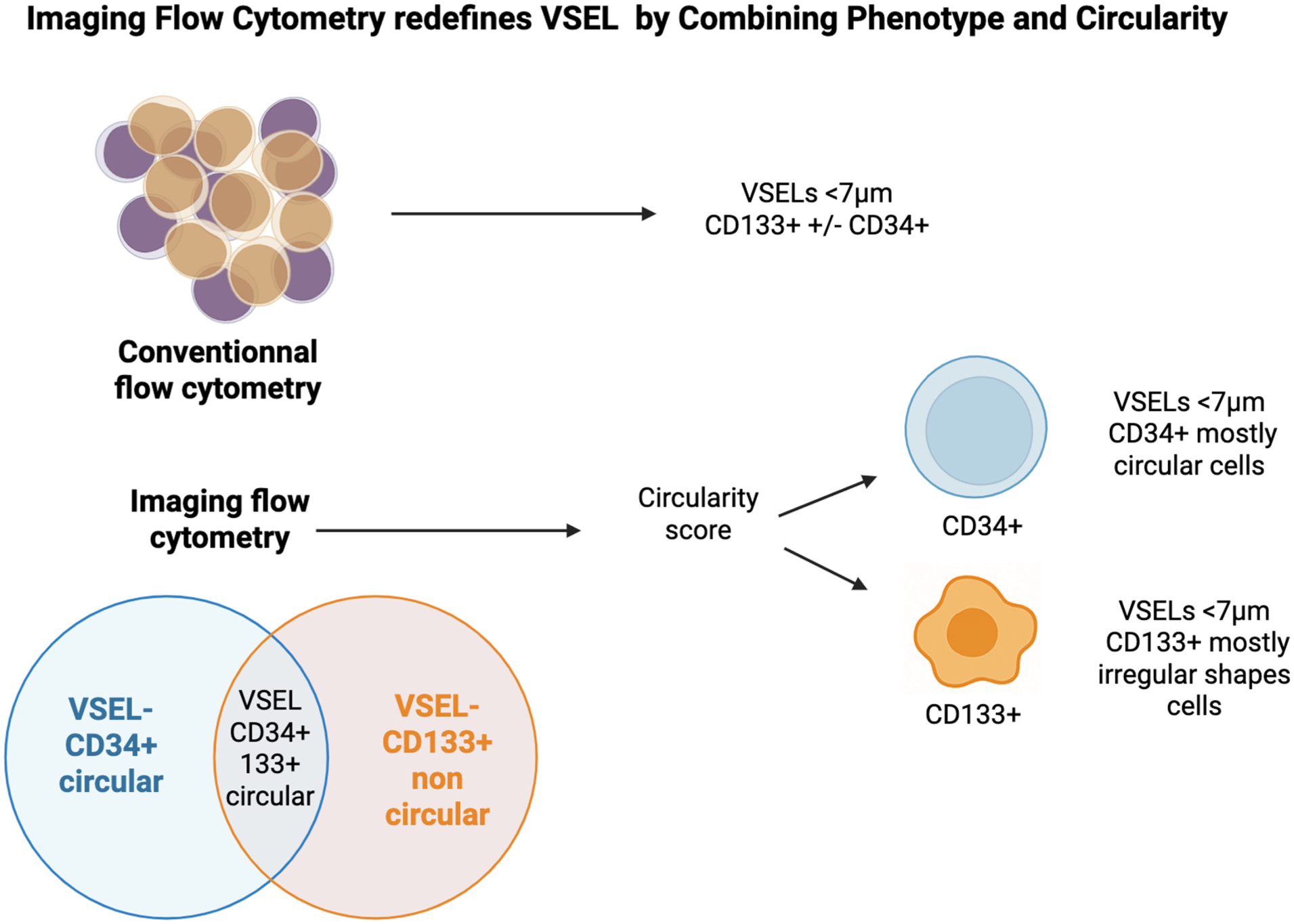
Abstract
Very small embryonic-like stem cells (VSELs) have been identified as potential precursors of the endothelial lineage in humans. Traditionally characterized by CD34 and CD133 markers, these populations remain poorly delineated, particularly regarding their morphological traits. Leveraging recent advances in imaging flow cytometry (iFC), this study sought to refine the characterization of VSELs by analyzing CD34+ and CD133+ Lin⁻ CD45⁻ cells from various sources, including bone marrow (BM), mobilized blood (MB), peripheral blood (PB), and cord blood (CB). Using high-resolution iFC, we assessed cell circularity and size, identifying significant morphological differences within subpopulations smaller than 7 µm. CD133+ cells predominantly exhibited irregular shapes, whereas CD34+ cells, regardless of CD133 co-expression, were mostly circular. Quantification across multiple sources revealed an enrichment of small circular CD34+ cells, especially in BM, MB, and CB. A comparative analysis with conventional flow cytometry (cFC) showed similar counts for CD34+ cells between methods (P = 0.06) but a significantly higher number of CD133+ cells with cFC (P < 0.0001), reflecting its inability to assess morphology. These findings highlight iFC as a powerful tool for rare stem cell analysis and propose CD34, rather than CD133, as a more reliable marker for identifying circular VSELs under 7 µm, refining future stem cell–isolation strategies.
Introduction
Very small embryonic-like stem cells (VSELs) are a unique class of primitive stem cells believed to be distributed across various postnatal human and murine tissues, including the bone marrow (BM), during early development1,2. These cells have also been identified in peripheral blood (PB), where their levels rise in response to physiological stress and tissue or organ damage3–5. Research suggests that these cells exhibit a broad capacity for differentiation across multiple germ layers, potentially giving rise to diverse tissue-specific stem cell populations. The existence of VSELs in postnatal murine and human tissues has been validated by multiple independent studies, challenging conventional perspectives on the hierarchy of adult stem cell compartments6,7. Evidence indicates that VSELs may differentiate into hematopoietic cells 8 , endothelial lineage4,9–12, or pulmonary alveolar epithelial cells13,14.
Initially discovered in humans as CD133 positive cells and nuclear OCT-415,16, VSELs also express CD34 17 . Our research has demonstrated the capacity of CD133+ VSELs 4 and also CD34+ VSELs 9 to differentiate into the endothelial lineage. As part of the development of CD34+ cells expanded in vitro—known as ProtheraCytes®—for use as a cell therapy in myocardial infarction, a robust and reproducible method was established to expand autologous CD34+ cells through an automated manufacturing process18–20. This process has been shown to enrich VSELs to approximately 5% 21 . Recent findings have shown that CD34+ VSELs exhibit fewer immaturity markers than CD133+ VSELs, as revealed by an mRNA analysis following conventional flow cytometry (cFC) isolation 22 , while they did not find any difference by a proteomic analysis. This paper proposes a potential hierarchy and interrelationship between CD34 and CD133 from VSELs to HSCs, combining morphology, size, and phenotype. Morphologically, VSELs have traditionally been described as round cells, predominantly identified in solid tissues such as the gonads23,24. However, objective quantification of this circularity, particularly in VSELs derived from blood or BM, has not previously been performed using non-subjective analytical tools.
Our study aims to accurately assess the morphology of CD34+ VSELs and CD133+ VSELs by analyzing their size and phenotype using imaging flow cytometry (iFC). This advanced imaging technology enables a deeper understanding of the structural properties of these stem cell populations. A key morphological parameter we will focus on is cell circularity.
Materials and methods
Patients
Human BM (n = 3), mobilized peripheral blood (MB; n = 5), and umbilical cord blood (CB; n = 5) samples were obtained from the Cell Therapy Unit of Saint-Louis Hospital (AP-HP, Paris; authorization number AC-2016-2759). PB (n = 5) samples were provided by the French Blood Bank Institute from informed healthy donors, in accordance with an agreement with Paris Descartes University (C CPSL UNT N 12/EFS/064). Peripheral venous blood (4 mL) was collected in plastic ethylenediaminetetraacetic acid (EDTA)-anticoagulated tubes and processed within 4 hours. The PB samples were diluted 1:1 in phosphate-buffered saline (PBS) containing 1% fetal bovine serum (FBS), while BM samples were diluted 1:4 in the same buffer. Mononuclear cells (MNCs) were isolated by centrifugation on a 1.077-g/mL Ficoll density gradient. After washing, MNCs were resuspended in PBS/1% FBS at a final density of 10 × 106 cells per mL and immediately processed.
Conventional and iFC staining
Samples were thawed slowly using Fluorescence-Activated Cell Sorting (FACS) buffer (PBS, 10% FBS, 2mM EDTA) previously heated at 37°C. After centrifugation (1260 × g, 6 min, acceleration at 7, deceleration at 3, 20°C), samples were resuspended in FACS buffer for counting. Antibodies were titrated, and cells were adjusted after counting at 5 million for the samples and 500,000 for monocolors and fluorescence minus one control. Staining was performed at 4°C for 30 min, and cells were stained using the following anti-human antibodies: BV421-conjugated anti-CD45 (clone HI30; BioLegend, San Diego, CA, USA), BV510-conjugated anti-CD14 (clone M5E2; BioLegend), BV605-conjugated anti-CD19 (clone HIB19; BioLegend), Pe-Cy7-conjugated anti-CD34 (clone 581; BioLegend), Allophycocyanin (APC)-conjugated anti-CD133 (clone AC133; Miltenyi Biotec, Bergisch Gladbach, Germany). For lineage staining, anti-human antibodies coupled to Fluorescein Isothiocyanate (FITC) were anti-CD235a (clone HIR2; BioLegend), anti-CD2 (clone RPA-2.10; BD Biosciences, Becton, Dickinson and Company, Franklin Lakes, NJ, USA), anti-CD3 (clone UCHT1; BioLegend), anti-CD16 (clone 3G8; BioLegend), anti-CD24 (clone ML5; BioLegend), anti-CD56 (clone 5.1H11; BioLegend), and anti-CD66b (clone G10F5; BioLegend). Dead cells were excluded using APC-Cy7-conjugated Zombie NiR (423106; BioLegend). Cells were then washed using FACS buffer before resuspension.
iFC acquisition and analysis
Acquisitions were performed on ImageStream®X MKII with the INSPIRE® software (Luminex Corporation, Austin, TX, USA) as previously described 25 . Acquisition was set for detecting lineage population on Ch02 with 488-nm laser (100 mW); CD34 on Ch06 with 561-nm laser (200 mw); Ch07 for CD45, Ch08 for CD19, and Ch10 for CD14 with 405-nm laser (120 mw); and finally CD133 on Ch11 and live/dead on Ch12 with 642-nm laser (150 mW). Ch1 and Ch9 were used to collet brightfield images on each camera. iFC data were analyzed using the IDEAS® software (Luminex Corporation, Austin, TX, USA). Cells were first selected by focus using the Gradient_RMS feature of Ch01 (“Focus”), and “Singlets” were isolated using Ch01 Area and Aspect Ratio features. We excluded dead cells and lineage positive cells (“Lin− Live”), and then focused on the “CD19− CD14− population. We analyzed in one hand the “CD133+ CD45+” population and then CD34 intensity (“CD34−” and “CD34+”); on the other hand, we looked at “CD34+ CD45−” cells and then at CD133 intensity (“CD133−” and “CD133+”). In both cases, we then apply a circularity feature on Ch01 and separate “non-circular” elements from “circular” ones. Once circular events selected, we isolated cells by size using Ch01 Diameter (“Diameter Low” and “Diameter High”). The circularity feature quantifies how much a mask’s shape deviates from a perfect circle by comparing the average distance of the object’s boundary from its center to the variation of these distances 26 . Objects that closely resemble a circle exhibit low variation and therefore yield high circularity values, while those with irregular shapes show higher variation and lower circularity values. Based on the morphological assessment of brightfield images, a threshold circularity score of 15 was established to differentiate between round and non-round cells, enabling consistent classification according to shape.
Method for quantifying VSELs per million CD45+ cells
Quantity of CD133+ and/or CD34+ cells is expressed per million of CD45+ cells to normalize the data independently of the cytometer and gating strategy used. CD45 is a common leukocyte marker present on all nucleated hematopoietic cells, providing a stable reference population. By relating CD133+ and/or CD34+ cells to the cells/singlets/live/CD45+ population, we minimize variability introduced by differences in gating strategies. This normalization approach ensures that results are comparable across different experimental cytometry platforms, allowing for more reliable interpretation and reproducibility of the data.
cFC analysis
Acquisitions were performed on an LSR Fortessa SORP (BD Biosciences, A division of Becton, Dickinson and Company, San Jose, CA, USA). Flow cytometry data were analyzed using FlowJo software. Live cells were isolated using Zombie Nir–negative population. Lineage-negative and CD14-negative CD19-negative cells were selected and then separated analyzed for CD45-negative CD34/CD133. The relative size of the cells was assessed in forward scatter (FSC) by using the Invitrogen Flow Cytometry Size Calibration Kit (#F13838) which contains a set of nonfluorescent microsphere suspensions reliable for size references (2, 4, 6, 10, and 15 µm).
Initial gating was performed on SSC-A vs FSC-A (gate A) to select the total cell population for analysis. From this population, singlets were selected based on FSC-A and SSC-A aspect ratios, and viable cells were gated using the Zombie NiR dye. All subsequent gates were derived from this parent population.
Statistical analysis
Statistical analyses were conducted using the non-parametric Mann–Whitney test or ANOVA, followed by Fisher’s protected least-significant difference test. A P-value of <0.05 was considered statistically significant. All analyses were performed using GraphPad Prism 5 (GraphPad Software Inc., San Diego, CA, USA) and the StatView software package (SAS, Cary, NC, USA).
Results
Viable BM Lin⁻ CD45⁻ 133+ cells are primarily non-circular cells in contrast to Lin⁻ CD45⁻ 34+
Both CD34+ VSELs and CD133+ VSELs have been described in the literature, but no study has systematically compared the different phenotypes of these cells. Then, we analyzed their expression and co-expression profiles in terms of CD34+ or CD133+ selection. Using MNCs isolated from BM samples of healthy subjects, we assessed the expression and/or co-expression of these markers within the same sample in BM. Events larger than 7 µm were excluded, and we specifically gated on Lin⁻ cells, including CD19⁻, CD14⁻, and CD45⁻ populations (Fig. 1). As shown in Fig. 1j, we observed three distinct phenotypes: CD34+ CD133⁻ (1339 ± 528 cells/millions of CD45+), CD34+ CD133+ (144 ± 62 cells/millions of CD45+), and CD133+ CD34⁻ cells (748 ± 543 cells/millions of CD45+).
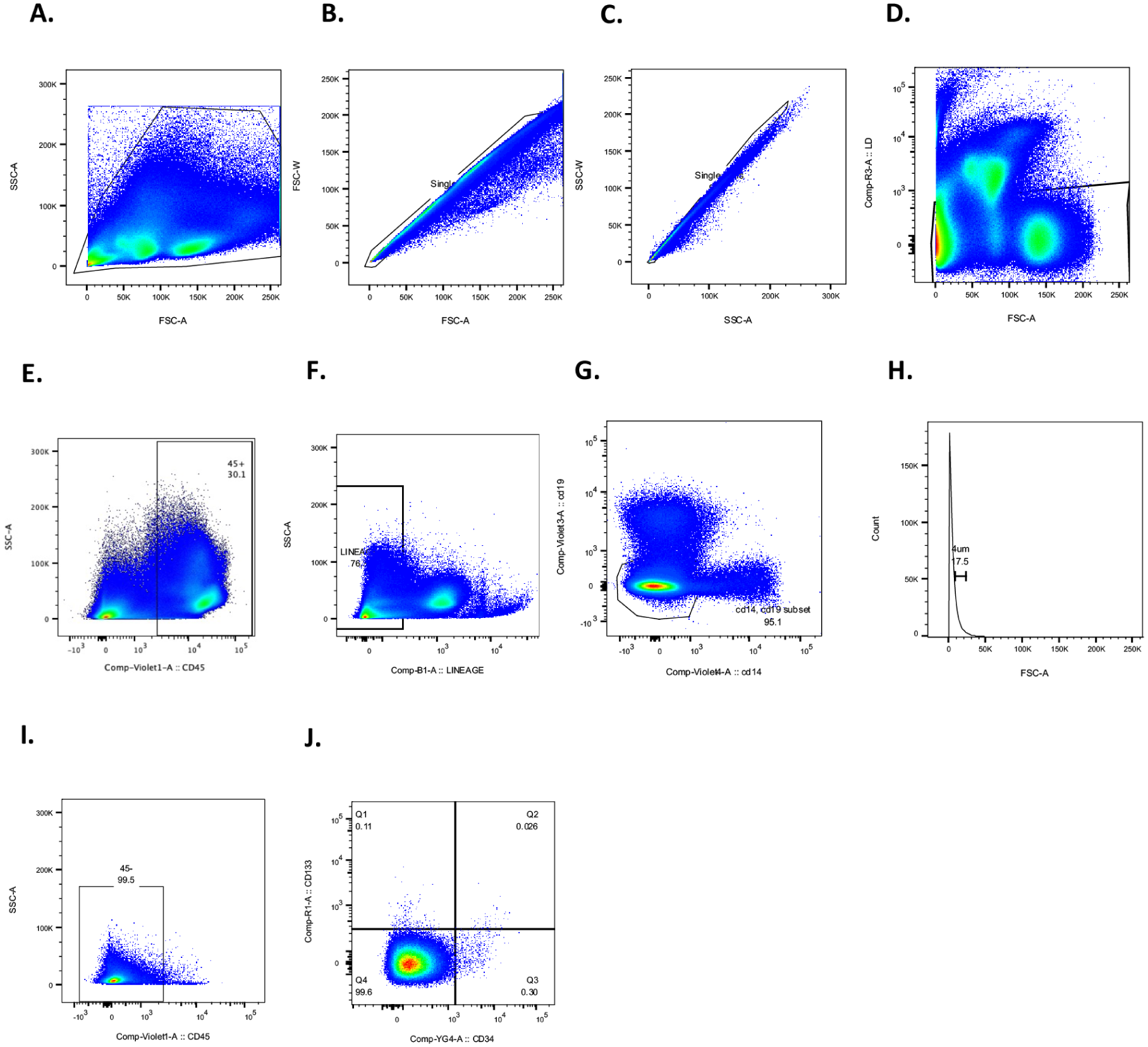
Gating strategy for the identification and quantification of VSELs among CD45+ cells by conventional flow cytometry. (a) Shows the initial SSC-A vs FSC-A plot used to gate total events. This gate served as the starting population for singlet, live cell, and lineage-negative gating shown in subsequent panels. Bone marrow (BM) cells were first analyzed using forward scatter (FSC) versus side scatter (SSC) to assess overall cell distribution. (b) Singlets were selected to exclude doublets and aggregates on FSC parameters. (c) Singlets were selected to exclude doublets and aggregates on SSC parameters. (d) From singlets gates, dead cells were excluded by selecting Zombie NiR (APC-Cy7) negative cells. (e) From the live cell gate defined in panel D, CD45+ cells were identified to enumerate total leukocytes, serving as an internal reference population. This gate was used exclusively for quantifying the absolute number of CD45+ events, enabling normalization of VSELs counts across samples. (f) From live gate in D, hematopoietic lineage-negative (Lin⁻) cells were defined as negative for FITC-conjugated antibodies targeting CD2, CD3, CD16, CD24, CD56, CD66b, and CD235a. (g) From lineage-gate in F, CD14⁻ and CD19⁻ populations were specifically highlighted to reinforce the exclusion of monocytes and B cells. (h) From the CD14− CD19− gate in G, events between 4 and 7 were selected using size calibration beads as reference standards. (i) Within the size range of interest population from H, CD45⁻ cells were further gated. (j) Among CD45⁻ gates from I, VSELs (Lin⁻ CD45⁻) were classified into three distinct subsets based on CD34 and CD133 expression: CD34+ CD133⁻, CD34+ CD133+, and CD133+ CD34⁻.
Thus, we decided to further investigate by applying the classical methods used for VSEL identification, adapting them to image cytometry to assess cell morphology alongside immunophenotype. Cells from the same samples were sequentially selected first based on CD133 or CD34 expression.
First, CD133+ VSELs were defined as live cells with a lineage-negative (Lin⁻), CD45⁻ (Fig. 2a). Among the CD133+ VSELs, we can distinguish two subpopulations, one CD34− (Fig. 2b) and another CD34+ (Fig. 2c). Among each, cells could be circular (circularity score above 15) or non-circular (circularity score below 15). CD45− CD133+ CD34−/+ can then be applied on populations selected on circularity features as illustrated in Fig. 2d, and proportions can be quantified based on this criterium (Fig. 2e) showing that CD133+ CD34⁻ cells predominantly (69%) exhibited a non-circular morphology, in contrast to CD133+ CD34+ cells, which were mostly (79%) circular (Fig. 2b, c).
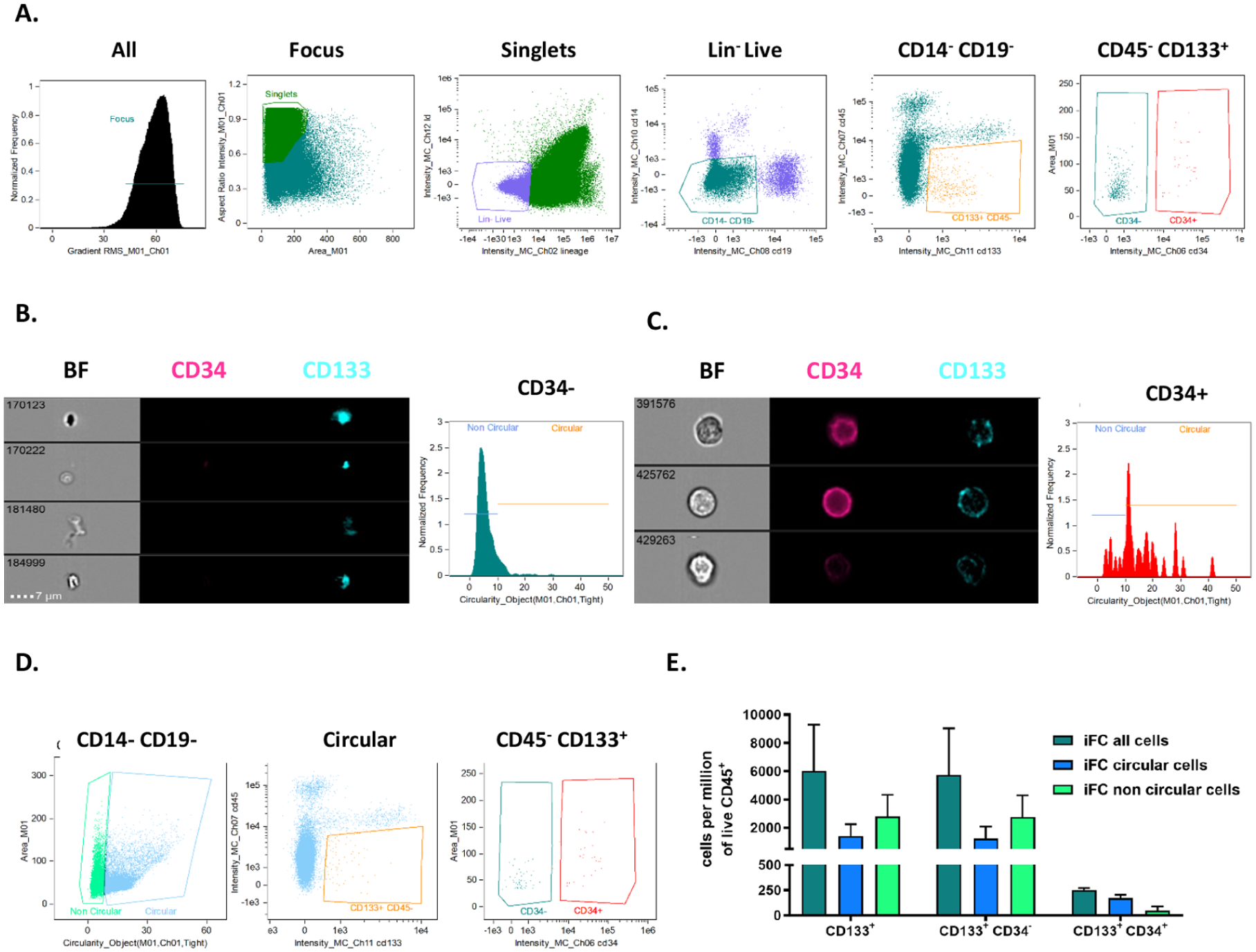
Characterization of bone marrow lineage⁻ CD45⁻ CD133+ cells by imaging cytometry. (a) Gating strategy of CD45⁻ CD133+ CD34⁻/+ after selection of focused live singlets, lineage-negative CD14⁻ CD19⁻ cells. (b) Images of Brightfield (BF), CD34 (pink), CD133 (blue), and circularity of lineage⁻ CD45⁻ CD133+ CD34⁻ cells. (c) Images of Brightfield (BF), CD34 (pink), CD133 (blue), and circularity of lineage⁻ CD45⁻ CD133+ CD34+ cells. (d) Circular cells were selected to distinguish morphologically round cells from irregular ones within the CD133+ CD34⁻ and CD133+ CD34+ subsets. (e) Quantification of circularity among CD133+ cells reveals that 69% of CD133+ CD34⁻ cells exhibit a non-circular morphology, while 79% of CD133+ CD34+ cells are circular.
Conversely, when analyzing live cells with a Lin⁻, CD45⁻, CD34+ phenotype (Fig. 3a), ImageStream imaging analysis demonstrated that most Lin⁻ CD45⁻ CD34+ cells were CD133⁻ (Fig. 3b, c). However, the majority of these cells exhibited a circular morphology (Fig. 3d, e) of, respectively, 69% and 72% circular for CD34+ CD133− and CD34+ CD133+.
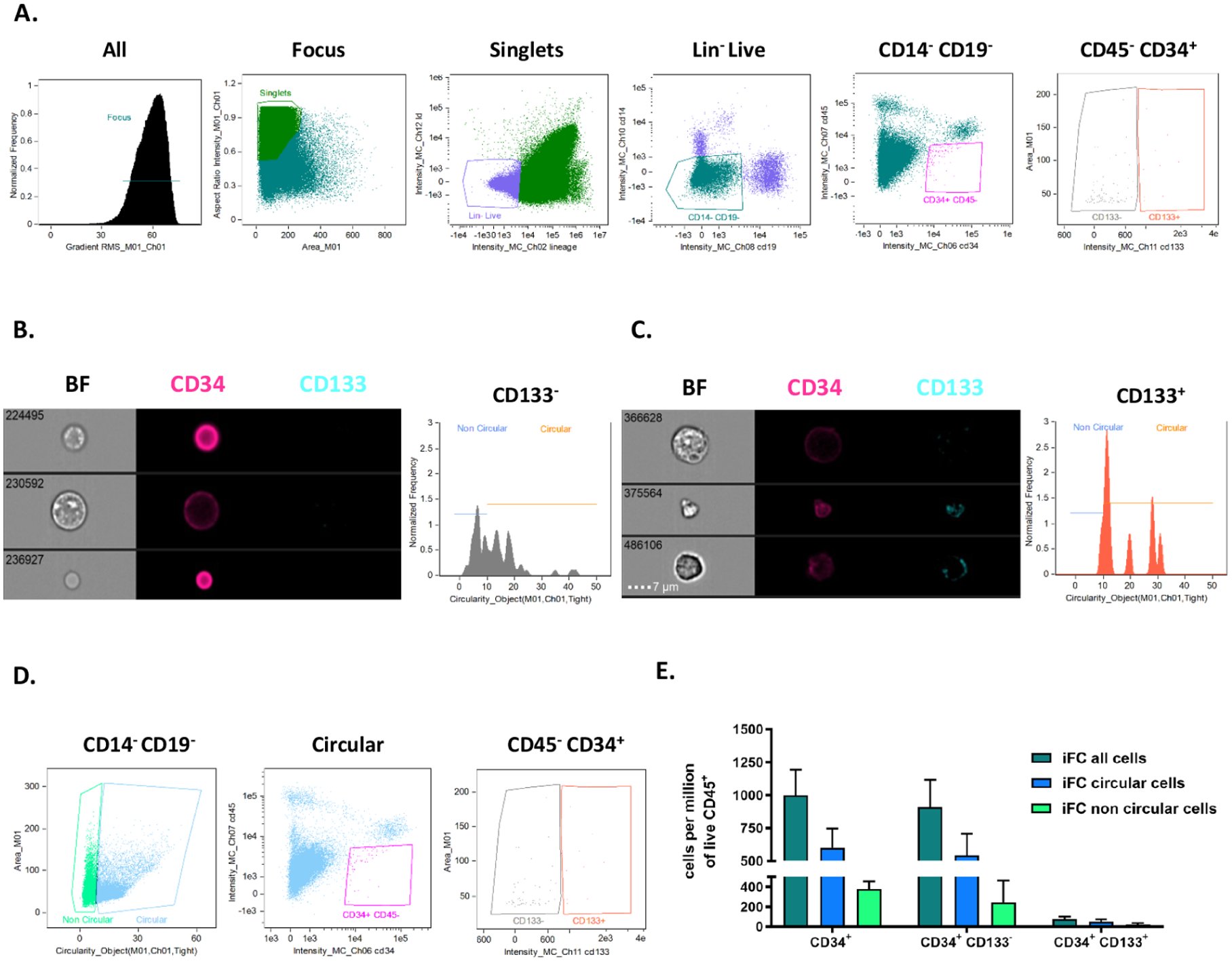
Bone marrow lineage⁻ CD45⁻ CD34+ cells by imaging cytometry. (a) Gating strategy of CD45⁻ CD34+ CD133⁻/+ after selection of focused live singlets, lineage-negative CD14⁻ CD19⁻ cells. (b) Images of Brightfield (BF), CD34 (pink), CD133 (blue), and circularity of lineage⁻ CD45⁻ CD34+ CD133⁻ cells. (c) Images of Brightfield (BF), CD34 (pink), CD133 (blue), and circularity of lineage⁻ CD45⁻ CD34+ CD133+ cells. (d) Circular cells were selected to distinguish morphologically round cells from irregular ones within the CD34+ CD133⁻ and CD34+ CD133+ subsets. (e) Quantification of circularity among CD34+ cells reveals that 69% of CD34+ CD133− cells exhibit a circular morphology, and 72% of CD34+ CD133+ cells are circular.
We then focused on circular Lin⁻ CD45⁻ CD34+ cells and analyzed their size, categorizing them as either with a diameter above or below 7 µm (Fig. 4a). Among cells with a diameter smaller than 7 µm, which meet the VSEL criteria, we identified (Fig. 4b) a significantly higher number of Lin⁻ CD45⁻ CD34+ CD133⁻ cells than Lin⁻ CD45⁻ CD34+ CD133+ cells. When normalized to the number of CD45+ cells within the initial MNC population, the mean count (± standard deviation) of small-sized CD34+ CD133⁻ cells were 502 ± 129 per million CD45+ cells. In contrast, the small-sized CD34+ CD133+ population was significantly lower, with an average count of 25 ± 2 cells per million CD45+. For larger cells (diameter > 7 µm), no significant difference was observed in the number of CD34+ CD133+ and CD34+ CD133⁻ cells (Fig. 4b). These findings suggest that the most prevalent circulating VSELs, as analyzed by iFC, are the CD34+ CD133− subset.

Differences in circular bone marrow lineage⁻ CD45⁻ CD34+ cells by size. (a) Selection of circular events prior to the gating of CD45⁻ CD34+ CD133⁻/+ cells and related shape in Brightfield (BF), ranked by size (< or >7 µm). (b) Quantification of circular bone marrow lineage⁻ CD45⁻ CD34+ CD133⁻/+ cell number per million CD45+ obtained by iFC.
cFC quantifies VSEL+ CD34+ more accurately than VSEL+ CD133+ compared to iFC across all compartments: BM, mobilized and PB, and CB
For analyzing rare stem cell populations, cFC is more commonly utilized due to its efficiency and reproducibility, so it is important to know what is the best technology to use for VSEL quantification or isolation. Fig. 5 provides a comprehensive quantification of circular Lin⁻ CD45⁻ CD34+ with a diameter <7 µm and >7 µm across different biological compartments with iFC and cFC. Fig. 5a,b illustrates the quantification of Lin⁻ CD45⁻ CD34+ negative or positive for CD133+ using iFC across four biological compartments: BM, mobilized blood (MB), PB, and CB. As expected, the results demonstrate that BM, MB, and CB harbor significantly higher numbers of Lin⁻ CD45⁻ CD34+ irrespective of their size or expression of CD133 than PB, confirming the well-established role of these compartments as primary niches for stem cells. Fig. 5c,d presents the quantification of the same cell populations using cFC. Importantly, Fig. 5d confirms that cFC detects a similar profile to iFC for the four different cellular compartments. Fig. 5e highlights key differences in the quantification of CD34+ and CD133+ VSELs between iFC and cFC. The quantification of CD34+ VSELs (Lin⁻ CD45⁻ CD34+) showed no significant difference between iFC and cFC. The mean and standard deviation values were 707 ± 373 in iFC compared to 1005 ± 802 in cFC (P = not significant). In contrast, the quantification of CD133+ VSELs (Lin⁻ CD45⁻ CD133+) revealed a significant difference between the two methods. The mean and standard deviation values were 509 ± 342 in iFC, whereas cFC yielded significantly higher values, with a mean of 3586 ± 1593. These findings suggest that while iFC and cFC produce comparable results for CD34+ VSELs, cFC tends to yield a much higher count for CD133+ VSELs, indicating potential differences in detection sensitivity or gating strategies between the two methodologies, aligning with the predominance of circular cells in the CD133+ VSEL subset.

Quantification of lineage⁻ CD45⁻ CD34+ cells by imaging and conventional flow cytometry (cFC) in four different compartments. (a) Analysis of Lin− CD45⁻ CD34+ CD133⁻/+ cells by iFC in bone marrow (BM), G-CSF-mobilized blood (MB), peripheral blood (PB), and cord blood (CB) from healthy donors. Related shape in Brightfield, ranked by size (< or > 7 µm), Illustrations are extracted from a BM sample. (b) Quantification of Lin− CD45⁻ CD34+ CD133⁻/+ cells by iFC in bone marrow (BM), G-CSF-mobilized blood (MB), PB, and CB from healthy donors. (c) Analysis of Lin− CD45⁻ CD34+ CD133⁻/+ cells by cFC in bone marrow (BM), G-CSF-mobilized blood (MB), PB, and CB from healthy donors. All events exceeding 2 μm are considered following a comparison with six bead particles of known standard diameters: 2, 4, 6, 10, and 15 μm. Illustrations are extracted from a BM sample. (d) Quantification of Lin− CD45⁻ CD34+ CD133⁻/+ cells by cFC in bone marrow (BM), G-CSF-mobilized blood (MB), PB, and CB from healthy donors. (e) Comparison of iFC and cFC methods for CD34+ VSELs—that is, <7 µm Lineage⁻ CD45⁻ CD34+—or <7 µm CD133+ VSELs—that is, <7 µm Lineage⁻ CD45⁻ CD133+ populations in the four compartments from healthy donors using a t-test (ns = non-significant, ***P < 0.0001).
Discussion
Our study provides novel insights into the morphological heterogeneity of VSELs by demonstrating, for the first time, that circularity—a previously underexplored parameter—serves as a meaningful discriminator between subpopulations defined by CD34 and CD133 expression. Using iFC, we observed that CD34+ VSELs are predominantly circular regardless of CD133 co-expression, while CD133+ VSELs, particularly those lacking CD34 expression, display a markedly non-circular morphology. This structural dichotomy, which has not been quantitatively assessed in prior studies, highlights circularity as a biologically relevant feature rather than a trivial morphological variation. Notably, primary gating on CD133 revealed that the majority of identified VSELs are non-circular, and a further sub-analysis showed that CD133+ CD34⁻ cells are more frequently non-circular, whereas CD133+ CD34+ cells tend to be circular. Conversely, CD34-based gating consistently retrieved predominantly circular cells, irrespective of their CD133 status. These findings emphasize that morphology, and specifically cell circularity, reflects a deeper layer of heterogeneity that may correspond to distinct functional or developmental stages within the VSEL compartment, thereby advancing our understanding of their classification and biological relevance.
The primary application of our research is to improve the isolation and classification of VSELs by integrating morphological parameters—particularly circularity and size—into existing immunophenotypic strategies. This combined approach enhances the accuracy of VSEL quantification and characterization, which is essential for their future application in regenerative medicine, especially in cardiovascular repair. Furthermore, given the existing discrepancies in the literature over the years regarding the characterization and phenotype of VSELs, a deeper understanding of the potential heterogeneity among VSEL subtypes is crucial. By refining morphological profiling, our study contributes to addressing these inconsistencies and lays a foundation for harmonizing identification protocols across studies. This is particularly important as several translational strategies involving VSELs are moving toward clinical application. Notably, expanded CD34+ (ProtheraCytes®) containing VSELs are currently under investigation for their therapeutic potential in myocardial infarction18,21,27. ProtheraCytes® enriched in CD34+ VSELs but also other descriptions by Lica et al.28,29 describing that VSELs can emerge in enriched in vitro cultures support the notion of a dynamic VSEL-like phenotype. We highlight that such culture systems could be valuable for future functional assays or validation of morphological findings, underscoring the clinical relevance of accurate and consistent VSEL identification.
Circularity, a key morphological characteristic in cell biology, has been linked to various aspects of stem cell function, including their identification, differentiation potential, and biological behavior26,30. Recent research suggests that circularity is relevant in imaging-based stem cell detection and in the functional properties of stem cells26,31. Circularity analysis has been used in deep-learning-based morphological assessments to distinguish pluripotent stem cells from differentiated cells 30 . Circularity has also been studied in relation to pluripotency and differentiation potential in embryonic stem cells and iPSCs. Changes in circularity have been associated with stem cell migration, adhesion, and colony-formation potential in vitro. In the context of hematopoietic stem cells, circularity has been linked to mechanical properties, with highly circular cells often exhibiting higher plasticity and resistance to mechanical stress 31 . While it serves as a useful imaging-based discriminant parameter for identifying specific stem cell subsets, further research is needed to understand the exact relationship between cell shape, plasticity, and stem cell function. VSELs represent a rare and developmentally early population of stem cells found in postnatal tissues such as BM, PB, and umbilical CB but also tissues like heart2,32. Their pluripotent-like properties and potential to differentiate into cell types of all three germ layers have attracted significant interest for regenerative applications1,2,33. However, a lack of consensus on their phenotypic markers, morphology, and in vivo functionality has limited their translation. Our study contributes to resolving these ambiguities by examining VSELs through a multi-parametric lens that includes surface marker expression, size, and for the first time in this context, cell circularity as a morphological feature. Incorporating circularity as a criterion into VSEL classification provides more resolution in identifying subpopulations. Importantly, it also aligns with the longstanding debate over the consistency and reproducibility of VSEL isolation. Since VSELs were first characterized, doubts have been raised within the scientific community regarding the reliability of their detection and functional assessment. Researchers have pointed to variability in marker-based isolation and inconsistencies in gating protocols as major sources of concern when identifying such scarce cell populations34,35. Nevertheless, these challenges were later countered by two separate publications6,7, which attributed the discrepancies to technical faults in cell-sorting methods rather than flaws in the original concept, emphasizing that accurate protocol execution is critical for successful VSEL identification. Quantification of VSELs has also been limited by inconsistencies in normalization strategies. cFC approaches often report VSEL numbers per milliliter of blood or as a percentage of cells obtained after Ficoll-based separation. However, expressing VSELs per milliliter is problematic because it does not account for cell loss during pre-analytical steps, variations in sample handling, or dilution effects. Furthermore, reporting VSELs as a percentage of cells from the Ficoll or buffy coat fractions is inherently ambiguous—raising the question, percentage of what? Such measurements lack precision because the cellular composition of these fractions is highly variable and may exclude a significant portion of the VSEL population due to their unique density and size characteristics. To overcome these limitations, we introduce a new and biologically grounded method of normalization: quantifying VSELs in relation to one million CD45+ cells present in the original whole blood sample. CD45+ leukocytes serve as a robust and consistent internal reference, affected by variable recovery rates or cell loss during separation—thus helping to eliminate bias. This strategy circumvents the confusion associated with volume-based quantification and percentage-based denominators that depend on incomplete or biased cell subsets. It also resolves complications related to volume loss, which disproportionately affects rare and low-density populations like VSELs during density gradient centrifugation. This normalization method enhances the biological relevance and clinical interpretability of VSEL quantification, establishing a more standardized and transparent framework for future research.
Our findings add nuance to this debate by showing that relying solely on immunophenotyping may obscure real differences in morphology that affect quantification and potentially functional interpretations. For instance, non-circularity could lead to over-detection in cFC or misclassification in gating strategies, contributing to variability between studies. This point is especially important when comparing our findings to the work of Bujko et al. 22 who isolated CD34+ and CD133+ VSELs from umbilical CB and performed molecular and proteomic characterization. Their study found that CD133+ Lin− CD45− VSELs expressed higher levels of pluripotency-associated genes such as Oct-4 and Nanog and showed elevated expression of the homing receptor CXCR4. These transcriptional differences suggest a more primitive state for CD133-positive VSELs. However, Bujko et al. 22 also noted no significant proteomic differences between CD34-positive and CD133-positive VSELs, raising questions about the translational implications of transcript-level distinctions. CXCR4+ SSEA-4+ Oct-4+ cells have historically been used to define VSELs 36 , particularly in early studies conducted in both mice 36 and humans 17 . In murine models, the first VSEL population was identified within the Sca-1+ Lin⁻ CD45⁻ fraction, which is functionally equivalent to the CD34+ Lin⁻ CD45⁻ population in humans. These cells were characterized by their small size, expression of stemness markers such as SSEA-4 and Oct-4, and their high migratory capacity mediated by CXCR4 in response to SDF-1 gradients 36 . Subsequent human studies confirmed the presence of similarly small cells in CB expressing CXCR4+ SSEA-4+ Oct-4+, supporting the idea that this phenotype represents a hallmark of human VSELs 17 . In the original reports, human VSELs were shown to co-express both CD34 and CD133 17 . Later consensus papers typically defined human VSELs as small CD133+ Lin⁻ CD45⁻ cells 2 . However, several independent studies—including our own—have successfully isolated CD34+ VSELs from human tissues and demonstrated their pluripotency. Notably, we have shown that both CD133+ and CD34+ VSELs can undergo endothelial differentiation4,9, first from BM 4 and later from CB sources 9 . Given that both CD34+ and CD133+ cells have been used to define VSELs recently in the literature, we aimed to address these discrepancies by refining their morphological characterization. In a recent study by Bujko et al., 22 CXCR4, Oct-4, and Nanog expression were found in both CD133+ and CD34+ VSEL subsets, although CD133+ cells tended to show slightly higher expression of certain pluripotency markers. These variations in reported phenotypes motivated us to better describe the morphological characteristics of CD34+ and CD133+ VSELs. Our study suggests that circular CD34+ CD133⁻ VSELs are more abundant, whereas non-circular CD133+ VSELs may represent a rarer but potentially more primitive subset, consistent with the findings of Bujko et al. Moreover, the reliability of CD133 as a stem cell and VSELs marker is subject to ongoing debate, and this issue merits careful consideration. As highlighted in a recent review 37 , CD133 (prominin-1) is not merely a passive marker of “stemness,” but a dynamic molecule whose expression and localization are regulated by the microenvironment, including hypoxic conditions and cellular stress. Its surface expression can fluctuate due to post-translational modifications or internalization, which may explain inconsistencies in detection across studies and tissues. Indeed, CD133 is not exclusively a surface marker. It has also been observed intracellularly, as shown in endothelial colony-forming cells (ECFCs) from human CB 38 , where its cytoplasmic localization was linked to enhanced vasculogenic function. A similar intracellular CD133 expression with functional implications has also been described in cancer cells39,40, where it contributes to aggressiveness or therapy resistance. These findings support the notion that CD133’s role is not limited to phenotypic identification but may also influence cellular function, including migration, differentiation, and vascular network formation. In the context of VSELs, while CD133+ Lin⁻ CD45⁻ cells have become a consensus phenotype in many studies, other markers such as CD34 are also used for isolation, and there remains heterogeneity in reported phenotypes.
In our study, we focused on both CD34+ and CD133+ VSEL populations and observed distinct morphological patterns between them. Our study helps bridge this gap by introducing morphology as a third dimension of classification—alongside immunophenotype and gene expression—which may account for the perceived inconsistencies in VSEL research. It is plausible that some of the difficulties in reliably reproducing VSEL isolation stem from the exclusion of non-circular or size-variable cells in sorting protocols. This reinforces the idea that circularity and size are not merely passive traits but potentially integral to defining VSEL identity and function.
Quantitatively, our iFC analysis identified that small-sized CD34+ CD133− cells (less than 7 µm) dominate the VSEL population, averaging over 500 cells per million CD45+ MNCs, while the CD34+ CD133+ subset was significantly rarer. These data support and extend those of Bujko et al. 22 who reported that CD34+ VSELs occur at approximately tenfold higher frequency than CD133+ VSELs in umbilical CB. The numerical dominance of CD34+ VSELs does not, however, undermine the importance of the CD133+ subset, which appears to represent a more stem-like, less differentiated state.
Technically, our findings also call attention to differences between iFC and cFC. While the two platforms yielded similar results for CD34+ VSELs, significant discrepancies arose in the quantification of CD133+ VSELs, with iFC detecting far fewer cells. This gap may be due to the exclusion of non-circular events in image analysis pipelines, inadvertently biasing results against morphologically irregular cells. Thus, our results highlight a critical need for refined gating strategies and morphologically aware cytometric analysis in the study of rare cell populations. Together, our work and that of Bujko et al. 22 support a layered model of VSEL heterogeneity. CD133+ VSELs, characterized by non-circular morphology, lower frequency, and high expression of pluripotency markers, appear to occupy a more primitive position in the stem cell hierarchy. CD34+ VSELs, being more circular and numerically dominant, may represent a more committed progenitor pool with greater clinical accessibility.
One limitation of this study lies in the interpretation of the non-circular CD133+ VSEL population. Due to their irregular morphology and very small size, distinguishing true intact cells from cellular debris, apoptotic bodies, or extracellular vesicles remains challenging, particularly when using iFC. This introduces uncertainty regarding the exact nature and viability of the CD133+ non-circular events. Furthermore, direct comparisons between iFC and cFC are constrained by inherent differences in the detection principles of the two platforms. Another key limitation is the absence of molecular expression of pluripotency-associated markers and also functional assays to determine whether the observed morphological differences according to circularity between CD34+ and CD133+ VSELs correspond to divergent differentiation potentials. Without lineage tracing or in vitro specification experiments, it remains unclear whether morphology reflects developmental hierarchy, functional state, or simply phenotypic variability. Future studies incorporating viability assessment, single-cell functional assays, and transcriptomic profiling will be essential to resolve these uncertainties and validate the biological significance of the morphological heterogeneity observed. cFC remains a cornerstone method in the accurate identification and quantification of rare cell populations such as VSELs. Its strength lies in the ability to perform high-throughput, multiparametric analysis with rigorous gating strategies, including essential tri-cellular discrimination—separating nucleated cells from debris, doublets, and anucleated elements based on scatter properties and nuclear staining. This level of resolution is critical for confidently isolating rare events like VSELs, which often fall near the analytical threshold. As a well-established, reference-standard technique, cFC offers a robust and reproducible platform that is widely validated across laboratories. In contrast, iFC, while useful for visual confirmation and spatial analysis, lacks the same level of standardization and statistical power, particularly for low-frequency events. It is therefore essential to recognize both the advantages and limitations of cFC. While it may miss certain morphological subtleties, it provides unmatched reliability when properly calibrated, with clearly defined gating strategies, fluorescence compensation, and internal controls. Understanding these methodological strengths—and their boundaries—is crucial to interpreting results accurately and avoiding both false positives and underestimations. As such, cFC remains an indispensable reference method, particularly when establishing benchmarks for novel quantification strategies or validating imaging-based technologies. These differences underscore the importance of choosing the appropriate VSEL subset for therapeutic applications, depending on whether pluripotency or practicality is prioritized. Moving forward, understanding how environmental and donor-specific factors such as delivery mode, sex, or systemic stressors affect VSEL morphology and marker expression will be critical. Moreover, future studies incorporating single-cell transcriptomics and epigenetic profiling, coupled with morphological analysis, may provide a more complete picture of VSELs’ identity and function. This will not only advance basic science but also improve the reproducibility and safety of VSEL-based regenerative therapies. As a final limitation, while the main focus of this study was on the CD34+ and CD133+ VSEL populations, we acknowledge the presence of a CD34– CD133– (“double-negative”) fraction within the Lin– CD45– gate. Although we applied the Zombie dye to exclude non-viable events, it remains possible that this fraction contains early apoptotic bodies, debris, or lineage-committed cells that may retain transient membrane integrity and thus escape detection by conventional viability markers. Previous studies have shown that early-stage apoptotic cells may not stain positively with DNA-binding dyes like 7-AAD, despite compromised viability [https://doi.org/10.1089/152581602321080619]. Therefore, the existence of a small subset of viable but poorly differentiated or phenotypically naïve cells within this double-negative gate cannot be completely excluded. This methodological limitation should be considered when interpreting the nature of these events, and future studies incorporating additional apoptotic markers and molecular profiling may help resolve their identity.
Conclusion
All in all, our study reveals that cell circularity is not a trivial morphological trait but a decisive marker: CD34+ VSELs emerge as structurally consistent and clinically accessible, while CD133+ cells display irregularity suggestive of deeper heterogeneity. By introducing CD45+-based normalization, we eliminate longstanding ambiguities in quantification and offer a robust framework for rare cell analysis. These findings challenge traditional gating dogmas and call for a paradigm shift—toward an integrated, morpho-phenotypic, and functional approach to VSEL characterization. Thus, this refined view opens the door to reproducible, standardized, and ultimately translatable VSELs isolation procedures. The next frontier lies in combining these insights with single-cell omics and functional validation to finally unlock the full regenerative promise of VSELs.
Footnotes
Acknowledgements
We thank all technicians in Saint Louis’ Hospital and in curie research center for their help.
ORCID iDs
Ethical Considerations
IRB Name / Approving Body: Ministère de l’Enseignement supérieur et de la Recherche and Agence Régionale de Santé Île-de-France | Approval Number / ID: AC-2022-5325 | Date of Approval: 8 November 2022.
Consent Statement
Human samples were obtained from the Cell therapy Unit of Saint-Louis Hospital (responsible authorities from cell therapy unit: Pr Larghero, AP-HP, Paris, authorization number AC-2022-5325 delivered on December 19th of 2022). All mothers (for cord blood) or patients provided written informed consent before enrollment. All mothers (for cord blood) or patients provided written informed consent to have their data published.
Author Contributions
All the undersigning authors have substantially contributed to the paper. DMS and CG designed the present study and wrote the manuscript. DMS, CG, MJ, and AC performed and/or analyzed the data. All authors declare that the submitted work is original and has not been published before (neither in English nor in any other language) and that the work is not under consideration for publication elsewhere.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by grants from the Conny-Maeva Charitable Foundation and the PROMEX STIFTUNG FUR DIE FORSCHUNG foundation.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Data Availability Statement
Raw data and detailed experimental procedures are available upon request to the corresponding author.
Statement of Human and Animal Rights
This article does not contain any studies with human or animal subjects.
Statement of Informed Consent
Human biological samples were used in this study. All participants provided written informed consent for participation, sample use, and publication, in accordance with ethical guidelines and approved by the relevant institutional review board (IRB: Ministère de l’Enseignement supérieur et de la Recherche and Agence Régionale de Santé Île-de-France, Approval No. AC-2022-5325, approved on 8 November 2022).