
Abstract
Neurodegenerative dementias, including Alzheimer’s disease and vascular dementia, have long been viewed through a neuron-centric lens. However, growing evidence highlights the indispensable and multifaceted roles of glial cells, astrocytes, microglia, and oligodendrocytes in both the onset and progression of these disorders. While prior reviews have cataloged glial dysfunction in isolation, this review offers a novel, integrative framework that maps the interconnected roles of glial subtypes across molecular, cellular, and circuit-level pathology in dementia. We critically synthesize recent advances in single-cell RNA sequencing, spatial transcriptomics, and glial imaging to redefine glial heterogeneity and function in disease states. Special emphasis is placed on the dynamic cross talk between glial populations and the feedback loops that govern their dual roles in neuroprotection and neurodegeneration. Furthermore, we examine emerging therapeutic strategies targeting glial-specific pathways, including NF-κB, JAK/STAT, CSF1R, and TREM2 signaling, as well as remyelinating agents and stem cell–based interventions. By integrating glial biology with therapeutic innovation, this review positions glial cells not as supporting actors but as central regulators and potential gatekeepers of dementia pathogenesis and treatment.
Keywords
Introduction
Dementia is a broad clinical syndrome marked by a progressive decline in cognitive functions, such as memory, reasoning, language, and behavior, that significantly impairs daily living and independence. Alzheimer’s disease (AD) is the most prevalent form, followed by vascular dementia (VaD), frontotemporal dementia, and Lewy body dementia. According to the data from the World Health Organization (WHO), in 2019, over 55 million people had dementia globally, posing an estimated economic burden of 1.3 trillion dollars 1 .
Historically, dementia research has focused predominantly on neuronal dysfunction and hallmark proteinopathies, such as amyloid-beta (Aβ) plaques and tau tangles. However, this neuron-centric view has increasingly come under scrutiny as glial cells, including astrocytes, microglia, and oligodendrocytes, have emerged as critical and dynamic regulators of both brain health and neurodegenerative processes. Once considered passive support cells, glia are now understood to engage actively in neurotransmission, metabolic support, immune surveillance, and synaptic pruning.
Astrocytes modulate neurotransmitter homeostasis, maintain blood–brain barrier (BBB) integrity, and provide metabolic support2,3. Microglia, the brain’s innate immune sentinels, respond to environmental cues and toggle between protective and neurotoxic phenotypes4,5. Oligodendrocytes, by myelinating axons, sustain white matter structure and rapid neuronal signaling 6 . As this review will explore, dysfunction in any of these glial populations contributes to pathological cascades in dementia.
In AD, for example, reactive astrocytes exacerbate inflammation and disrupt neuronal support, while chronically activated microglia perpetuate cytokine-driven neurodegeneration. Oligodendrocyte loss and demyelination impair neural conduction and contribute to cognitive decline, especially in vascular-related pathology. Recent work has also emphasized the importance of inter-glial communication, revealing shared signaling hubs (e.g. NF-κB, JAK/STAT, TREM2, CX3CR1) that coordinate neuroimmune and neurodegenerative responses.
Despite these advances, most existing reviews tend to examine glial cell types in isolation or focus narrowly on specific mechanisms. Reviews such as Deng et al. 3 and Ferreira et al. 4 have provided valuable glia-centric overviews but often lack a unified systems-level model that captures glial dynamics across dementia subtypes and therapeutic modalities.
The current review addresses this gap by introducing an integrative framework that:
Maps the interconnected roles of astrocytes, microglia, and oligodendrocytes in dementia progression;
Synthesizes glial cross-talk mechanisms and their implications for neuroinflammation and neurodegeneration;
Highlights single-cell transcriptomic technologies redefining glial heterogeneity;
Critically evaluates glia-targeted therapies, spanning inflammation modulation, remyelination, and stem cell–based interventions.
Through this comprehensive approach, we reposition glial cells not as peripheral participants but as central architects of both dementia pathogenesis and its therapeutic future. By shifting our focus beyond neurons and into the dynamic world of glia, we may uncover novel intervention points that could revolutionize dementia treatment and management.
Glial cells in the healthy brain
Glial cells play indispensable roles in orchestrating neural function and maintaining cerebral homeostasis. Far from being mere neuronal support elements, astrocytes, microglia, and oligodendrocytes engage in complex, dynamic interactions with neurons, blood vessels, and each other. Their specialized functions extend beyond maintenance to include synaptic regulation, immune surveillance, and bioenergetic support, all of which are foundational to healthy brain function.
Astrocytes
Astrocytes are multifunctional glial cells that form an essential interface between neurons, blood vessels, and the extracellular environment. A critical yet often underappreciated function is their role in astrocyte-neuron lactate shuttling, whereby astrocytes metabolize glucose to lactate and deliver it to neurons as a rapid energy source during periods of heightened synaptic activity. This mechanism is crucial for sustaining long-term potentiation and memory formation 3 . Astrocytes also maintain glutamate homeostasis by clearing excess neurotransmitters through excitatory amino acid transporters (EAATs), thereby preventing excitotoxicity. Their perivascular endfeet regulate BBB integrity, modulate cerebral blood flow via vasoactive signaling, and help buffer extracellular potassium, further supporting synaptic fidelity 2 . These diverse functions position astrocytes as metabolic gatekeepers and synaptic modulators in the healthy brain.
Microglia
Microglia serve as the brain’s resident immune sentinels, continuously surveying the parenchyma through dynamic ramified processes. During early development, they mediate activity-dependent synaptic pruning, eliminating weak or redundant synapses via complement-mediated signaling (e.g. C1q, C3) to sculpt neural circuits 7 . This function continues in adulthood under homeostatic conditions, albeit with more selective surveillance. In the healthy brain, microglia also release trophic factors such as insulin-like growth factor 1 (IGF-1) and brain-derived neurotrophic factor (BDNF) to support neuronal survival, and play roles in oligodendrocyte maturation and myelin turnover. Importantly, microglia maintain a tightly regulated balance between immunosurveillance and inflammation; disruption of this balance predisposes the central nervous system (CNS) to chronic neuroinflammation and disease.
Oligodendrocytes
Oligodendrocytes are responsible for producing the myelin sheath that ensheathes axons, dramatically enhancing the conduction velocity of action potentials through saltatory transmission. Beyond insulation, oligodendrocytes provide metabolic support to axons by supplying lactate via monocarboxylate transporters (MCT1), which is especially vital in long-projecting neurons susceptible to metabolic stress 8 . Each oligodendrocyte can myelinate segments of multiple axons, forming a functional unit that supports white matter integrity and neural connectivity. Oligodendrocyte lineage cells also participate in adaptive myelination, where neuronal activity influences myelin remodeling, suggesting a bidirectional relationship between axons and glia in maintaining plasticity.
Figure 1 illustrates an overview of these three major glial cell types and their core physiological roles in maintaining a healthy CNS.
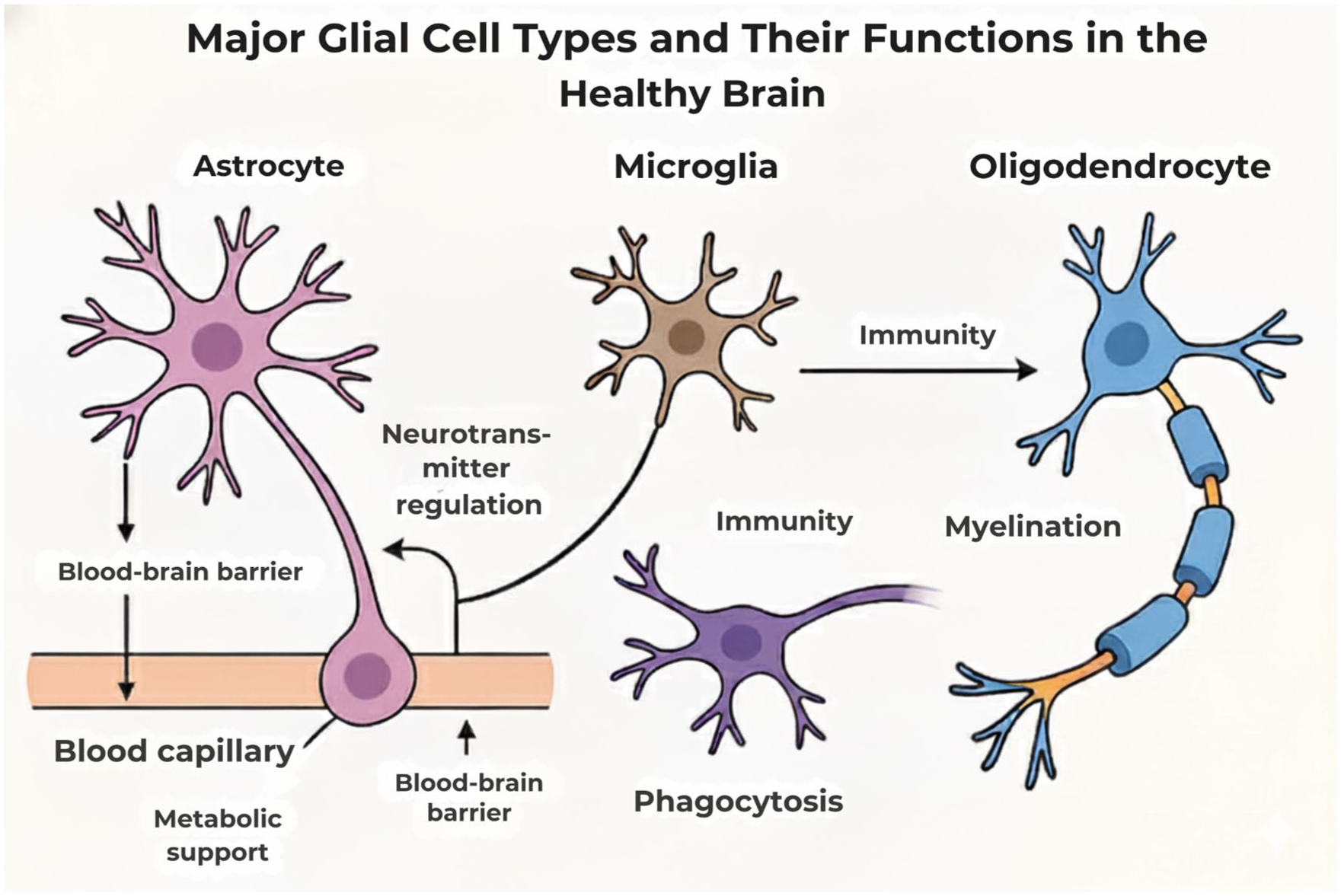
Major glial cell types and their functions in the healthy brain.
Glial dysfunction in AD
In AD, glial cells play an active, disease-driving role that extends far beyond merely responding to neuronal injury. The shift from homeostatic to reactive glial phenotypes, characterized by pro-inflammatory, phagocytic, and metabolic dysfunction, underpins many of the pathological processes in AD. Crucially, recent findings reveal that glial–glial signaling, rather than glial–neuron interactions alone, is a central feature of AD progression. Understanding how astrocytes, microglia, and oligodendrocytes interact and respond to evolving pathological cues opens new avenues for therapeutic targeting.
Figure 2 represents a summary of pathological transformations in astrocytes, microglia, and oligodendrocytes during AD, including key signaling pathways and emerging therapeutic targets.

Glial cell dysregulation in the pathophysiology of Alzheimer’s disease. Depicts alterations in glial function associated with Alzheimer’s disease, including microglial activation, astrocytic reactivity, impaired amyloid clearance, and neuroinflammatory signaling contributing to disease progression.
Astrocytes in AD
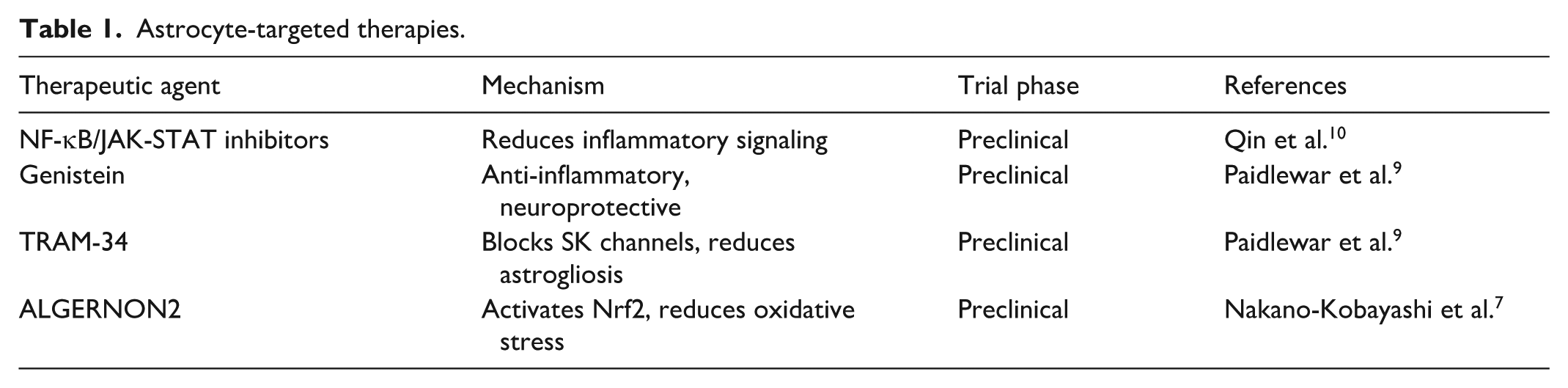
Astrocytes undergo profound transcriptional and morphological changes in response to Aβ and tau accumulation. Initially neuroprotective, these cells can enter a reactive state, often broadly classified as the A1 phenotype, which exacerbates neuroinflammation, impairs glutamate clearance, and compromises the BBB3,9. This transformation is orchestrated by pro-inflammatory pathways including NF-κB, JAK/STAT, and GPCR signaling. Importantly, complement C3, largely produced by reactive astrocytes, has emerged as a key mediator of glial toxicity. C3 interacts with the microglial complement receptor 3 (CR3) to promote synaptic engulfment and neuron loss, representing a crucial astrocyte–microglia signaling axis 10 . Preclinical studies suggest that interventions targeting this axis, such as C3 inhibitors or C3aR antagonists, may preserve synapses and attenuate inflammation. Other potential interventions explored in AD models include Nrf2 pathway activators (e.g. ALGERNON2) that reduce reactive astrocytosis and natural compounds like genistein, which exert anti-inflammatory effects and rescue synaptic function 11 .
Microglia in AD
Microglia exhibit a dynamic phenotypic shift in AD, transitioning from homeostatic surveillance to a disease-associated microglia (DAM) profile under the influence of Aβ and neuronal stress signals. While early activation may promote debris clearance, chronic stimulation leads to maladaptive inflammation, complement-mediated synaptic loss, and oxidative stress4,12. Two major regulatory hubs, triggering receptor expressed on myeloid cells 2 (TREM2) and CD33, modulate microglial fate in AD. TREM2 signaling generally enhances lipid metabolism, phagocytosis, and Aβ clearance, while CD33 activation can inhibit these functions and promote a pro-inflammatory state. Clinical strategies targeting these molecules have included colony-stimulating factor 1 receptor (CSF1R) inhibitors, which deplete activated microglia populations (which are often subsequently replaced by circulating cells), and glucagon-like peptide-1 (GLP-1) receptor agonists (e.g. NLY01) that show promise in suppressing inflammatory signaling in preclinical models 13 .
Beyond classical mediators, the fractalkine signaling axis (CX3CL1/CX3CR1) has emerged as a critical modulator of neuron–microglia and astrocyte–microglia interactions. Under physiological conditions, neuronal CX3CL1 suppresses microglial activation via CX3CR1. In AD models, disruption of this axis leads to exaggerated microglial reactivity and contributes to synaptic and myelin pathology 14 . This pathway also indirectly influences oligodendrocyte survival by altering the inflammatory milieu and oxidative burden.
Oligodendrocytes in AD
Although traditionally understudied in AD, oligodendrocytes are now recognized as active contributors to neurodegeneration. Myelin degradation and white matter loss are early features of AD, especially in regions critical for cognitive function. In AD, oligodendrocyte progenitor cells (OPCs) exhibit impaired maturation, while mature oligodendrocytes display immune-like phenotypes and reduced capacity for remyelination 6 . Microglia-derived cytokines, including IL-1β, TNF-α, and C1q, have been shown to disrupt oligodendrocyte function and promote myelin breakdown. In addition, astrocyte-microglial dysfunction indirectly hampers OPC proliferation and differentiation by altering metabolic and inflammatory support. Therapeutically, agents such as clemastine (a repurposed antihistamine) and benztropine are being investigated for their potential to promote remyelination by stimulating OPC differentiation. Anti-inflammatory compounds targeting the broader glial network may also help preserve oligodendrocyte integrity. Given the interconnectedness of glial signaling, combination therapies addressing multiple glial types may be required for effective intervention.
Glial cells in VaD
VaD, the second most common form of dementia globally, arises primarily from cerebrovascular pathology, such as chronic hypoperfusion, small vessel disease, and ischemic stroke, that disrupts brain connectivity and impairs cognitive function. Unlike AD, where proteinopathies dominate, VaD pathogenesis is tightly linked to neurovascular dysfunction. Within this framework, glial cells, especially astrocytes, microglia, and oligodendrocytes, play pivotal and increasingly appreciated roles. Their dysregulation contributes not only to neural damage but also to failed repair, highlighting them as therapeutic targets for disease modification.
Astrocytic dysfunction
Astrocytes form a vital interface between neurons and the cerebral vasculature. In VaD, these cells respond to cerebral hypoperfusion and ischemic insults by entering a reactive state. This can lead to the detachment of astrocytic endfeet from the vasculature, destabilizing the neurovascular unit and compromising BBB integrity 15 . The increased permeability permits neurotoxic plasma proteins, leukocytes, and oxidative agents to infiltrate the parenchyma, fueling neuroinflammation. Mechanistically, astrocytic dysfunction involves downregulation of tight junction-supporting proteins (e.g. claudin-5, occludin) and upregulation of aquaporin-4 (AQP4), which can exacerbate cerebral edema 16 . In addition, the loss of astrocyte-mediated lactate shuttling and K⁺ buffering worsens metabolic stress in ischemic regions. Therapeutic approaches being explored in animal models include BBB stabilizers (e.g. minocycline, resveratrol) and AQP4 modulators.
Microglial activation
In response to vascular pathology, microglia shift toward a chronically activated phenotype that contributes to white matter degeneration, a hallmark of VaD. Hypoxia and BBB breakdown trigger excessive microglial release of IL-1β, TNF-α, and IL-6, initiating a neuroinflammatory cascade that damages oligodendrocytes and disrupts axonal integrity17,18. Activated microglia also interfere with synaptic plasticity and repair by pruning viable synapses, a process thought to be mediated via complement pathways and fractalkine dysregulation. Preclinical models such as bilateral common carotid artery stenosis (BCAS) in mice are widely used to study these dynamics, replicating chronic cerebral hypoperfusion and microglial activation seen in human VaD. Therapeutic interventions under investigation in these models include P2X7 receptor antagonists and CX3CR1 modulators.
Oligodendrocyte loss and myelin degeneration
Oligodendrocytes are exceptionally vulnerable to ischemia and oxidative stress. In VaD, their degeneration contributes to myelin sheath disruption in deep white matter regions, particularly in periventricular areas and the corpus callosum. The ensuing conduction delays and loss of axonal support directly correlate with cognitive impairment. Recent studies show that OPCs are often present in VaD lesions but fail to mature, due to an inflammatory microenvironment and disrupted trophic support. Pro-inflammatory cytokines and glutamate excitotoxicity further inhibit remyelination. This demyelination impairs signal transmission and weakens synaptic connectivity, and the death of oligodendrocytes contributes to a loss of trophic support for axons, making them more vulnerable to degeneration 6 . Therapeutic strategies under investigation include remyelinating agents like clemastine fumarate and siponimod, as well as cell-based therapies involving transplantation of human OPCs, which have shown remyelination potential in ischemia-induced demyelination models.
Figure 3 summarizes the interplay of astrocytic, microglial, and oligodendrocytic dysfunctions in VaD, showing how ischemic and inflammatory events lead to BBB breakdown, white matter injury, and cognitive decline.

Glial contributions to the pathogenesis of vascular dementia. Highlights the involvement of glial cells in vascular dementia, focusing on blood–brain barrier disruption, neurovascular unit dysfunction, astrocytic end-feet detachment, and inflammatory cascades driven by glial dysregulation.
Inter-glial communication in dementia
The progression of both AD and VaD is not solely a consequence of isolated glial dysfunction; it is critically driven by disrupted communication between glial subtypes. In the healthy brain, astrocytes, microglia, and oligodendrocytes engage in a tightly regulated signaling network that sustains homeostasis, synaptic integrity, and metabolic balance. In disease states, however, this intricate dialogue becomes distorted, leading to a cascade of maladaptive responses that accelerate neuronal injury.
Figure 4 provides a schematic contrast between healthy glial communication and the disrupted pathways that emerge in neurodegeneration, highlighting key signaling mediators.

Inter-glial communication and cross talk in dementia. Visualizes dynamic interactions between astrocytes, microglia, and oligodendrocytes under pathological conditions, emphasizing the feedback loops and signaling networks that exacerbate neurodegeneration in various forms of dementia.
Molecular pathways mediating glial cross talk
Several key molecular pathways mediate both cooperative and antagonistic signaling among glia. When dysregulated, they can shift glial cells into neurotoxic phenotypes:
NF-κB Pathway: A central regulator of glial inflammation, NF-κB is activated in both astrocytes and microglia in response to Aβ, hypoxia, or cytokines. This pathway amplifies the production of pro-inflammatory mediators, fostering a feed-forward loop of glial activation.
TREM2 Signaling: Expressed on microglia, TREM2 modulates responses to lipid debris, Aβ, and dying cells. In healthy states, TREM2 promotes phagocytosis and anti-inflammatory microglial profiles. Impaired TREM2 signaling can shift microglia toward a neurotoxic phenotype, which in turn can stimulate astrocytic NF-κB activation.
Complement C3/C3aR Axis: Activated astrocytes upregulate complement component C3, which binds to microglial C3a receptors (C3aR), pushing microglia toward a neurodegenerative (MGnD) phenotype. This interaction can disrupt synaptic integrity and contribute to demyelination by promoting inflammation around oligodendrocytes.
CX3CL1-CX3CR1 Axis: Neurons and astrocytes release fractalkine (CX3CL1), which binds to CX3CR1 on microglia to regulate their surveillance. In neurodegeneration models, reduced CX3CL1 availability or impaired CX3CR1 signaling can lead to unchecked microglial activation, disrupting microglia-oligodendrocyte support and contributing to white matter loss.
SPP1 (Osteopontin): Recently recognized as a key mediator of glial cross talk, secreted phosphoprotein 1 (SPP1) is upregulated in reactive astrocytes and DAM. It influences glial metabolism and phagocytosis. While SPP1 may have reparative roles in acute injury, chronic overexpression is associated with fibrotic gliosis and impaired remyelination.
Cooperation versus conflict: dual nature of glial signaling
In early or compensatory phases of neurodegeneration, cooperative signaling helps mitigate damage: astrocytes release lactate, microglia clear debris, and CX3CL1-CX3CR1 interactions maintain a non-inflammatory state. However, in chronic disease, antagonistic signaling loops emerge: C3 release from A1 astrocytes activates C3aR on microglia; pro-inflammatory microglia release IL-1α, TNF-α, and C1q, converting astrocytes to neurotoxic A1 phenotypes; and oxidative stress from reactive glia inhibits OPC differentiation, impairing remyelination. These dysfunctional interactions create a hostile neural environment marked by synaptic loss, impaired remyelination, and cognitive decline.
Therapeutic implications
Given the centrality of inter-glial communication, emerging therapies aim to modulate rather than suppress glial signaling. Strategies under preclinical investigation include TREM2 agonists (e.g. AL002) to enhance microglial clearance, C3 inhibitors (e.g. compstatin analogs) to reduce harmful astrocyte-microglia signaling, and CX3CR1 enhancers to restore homeostatic microglial surveillance.
Glial cell heterogeneity and technological advances
The glial landscape of the brain is far from uniform. Recent advances in single-cell RNA sequencing (scRNA-seq) and spatial transcriptomics have revolutionized our understanding of glial diversity, revealing a complex tapestry of transcriptionally distinct subtypes that vary across brain regions, disease stages, and pathological contexts. These technologies allow high-resolution mapping of gene expression patterns at the single-cell level, shedding light on how glial cell states diverge in AD, VaD, and mixed dementias. Early views cast glia into broad functional categories (e.g. A1/A2 astrocytes), but current data dismantle this simplicity, suggesting instead that glial cells exist along continuums of activation and specialization.
Figure 5 represents this diversity, highlighting DAMs, reactive astrocyte subtypes, and oligodendrocyte lineage variability.

Heterogeneity of glial cell populations in dementia pathologies. Represents the diversity of glial subtypes and region-specific phenotypes in dementia, including disease-associated microglia (DAMs), A1/A2 reactive astrocytes, and oligodendrocyte lineage variability, highlighting their context-dependent roles in neurodegeneration.
Astrocyte heterogeneity
scRNA-seq has uncovered astrocyte subpopulations with region-specific transcriptional programs. In AD, the A1 (pro-inflammatory) and A2 (neurotrophic) classification has been a useful, though simplified, heuristic 5 . Newer classifications identify subsets involved in BBB maintenance, metabolic buffering, and synaptic pruning. Spatial transcriptomics further reveals that astrocyte phenotypes differ between the hippocampus, cortex, and subcortical white matter. Astrocytes near amyloid plaques, for example, express higher levels of complement genes (e.g. C3, SERPING1), suggesting localized neuroinflammatory specialization.
Microglial subpopulations
Microglia display striking transcriptional heterogeneity. scRNA-seq has identified several subtypes, including:
Homeostatic microglia, marked by P2RY12, TMEM119, and CX3CR1.
DAMs, expressing TREM2, Apoe, Cst7, and Lpl, with roles in Aβ clearance.
Pro-inflammatory microglia, enriched for IL1B, TNF, and NF-κB targets.
Interferon-responsive microglia, associated with synaptic loss 19 .
Spatial profiling has shown that DAMs localize preferentially to peri-plaque zones in AD, whereas microglia in white matter in VaD models exhibit gene signatures linked to ischemic stress and mitochondrial dysfunction.
Oligodendrocyte lineage complexity
OPCs also appear highly diverse. scRNA-seq studies have delineated multiple stages of maturation, with transcriptomic shifts in:
Myelin-related genes (e.g. MBP, PLP1) in mature oligodendrocytes.
Inflammation and ECM remodeling genes (e.g. SPP1, MMP9) in stressed OPCs.
Antigen-presentation genes (e.g. HLA-DR) in oligodendrocytes from chronic disease states.
In VaD models, white matter oligodendrocytes show a unique signature of hypoxic stress, with upregulation of HIF1A, CHOP, and pro-apoptotic factors.
Clinical implications
These high-resolution transcriptomic maps provide a molecular atlas of glial dysfunction. This enables the conceptualization of more precise therapeutic strategies, such as subtype-specific drug targeting (e.g. inhibiting C3+ astrocytes while preserving A2 subsets) and the development of early disease biomarkers (e.g. DAM signatures or OPC stress markers) detectable in CSF or via imaging.
The convergence of scRNA-seq, spatial transcriptomics, and epigenomic profiling is paving the way toward precision neuroimmunology, transforming glial cells from passive bystanders to central targets in dementia intervention strategies.
Emerging therapeutics targeting glial cells
Glial cells have emerged as compelling therapeutic targets in dementia due to their central roles in neuroinflammation, synaptic maintenance, and white matter integrity. This section discusses targeted strategies and highlights the critical distinction between preclinical findings and clinical applicability.
Astrocyte-targeted therapies
Astrocytes regulate BBB integrity, neurotransmitter cycling, and antioxidant defense. In dementia, they often adopt a reactive phenotype, exacerbating inflammation. Therapies explored in preclinical models aim to suppress this harmful transformation. These are summarized in Table 1.
Astrocyte-targeted therapies.
Microglia-targeted therapies
Microglia are vital for responding to injury and clearing debris. In neurodegeneration, their chronic activation can become neurotoxic. Therapeutic efforts aim to shift them toward a homeostatic, neuroprotective state. These strategies are summarized in Table 2.
Microglia-targeted therapies.

Oligodendrocyte-targeted strategies
Oligodendrocytes are essential for myelin production. Their degeneration contributes to cognitive decline, especially in VaD. Strategies to protect them or promote remyelination are summarized in Table 3.
Oligodendrocyte-targeted strategies.

Major barriers to clinical translation
While the strategies in Tables 1–3 show promise in simplified models, their translation to human dementia patients is fraught with challenges. This represents a significant knowledge gap, and reasons for failure often include:
Species Differences: Glial gene expression and immune responses in rodent models often fail to recapitulate human disease.
Delivery to the CNS: The BBB remains a formidable obstacle, preventing most drugs from reaching therapeutic concentrations in the brain.
Toxicity and Specificity: Many interventions (e.g. pathway inhibitors) are broad, and long-term suppression of inflammation or glial function can impair essential brain maintenance, healing, and immune surveillance.
Patient Heterogeneity: Dementia is a complex syndrome. A patient with mixed AD-VaD pathology may respond differently than one with pure AD, but clinical trials rarely stratify for these differences.
Clinical Trial Failures: The neurodegeneration field is marked by a high rate of clinical trial failures. This highlights a poor understanding of the correct therapeutic window (i.e. treating too late) and a lack of reliable biomarkers to measure target engagement and therapeutic effect.
Future directions
The expanding understanding of glial heterogeneity presents new research directions, but also reveals significant gaps in our knowledge. Future success will depend on moving beyond broad suppression toward fine-tuned modulation.
Unresolved questions and knowledge gaps
While descriptive content on glial activation is vast, the field lacks a deeper analytical synthesis. Key unresolved questions and limitations of current models must be addressed.
Limitations of Current Models: Are the A1/A2 astrocyte or DAM microglial phenotypes discrete, stable states, or are they transient points on a complex spectrum? Current in vitro and animal models often fail to capture this complexity or the chronic, progressive nature of human dementia.
Inconsistencies in Glial Phenotypes: Studies often report conflicting glial activation phenotypes. This may be due to differences in species (mouse vs human), disease stage, brain region, or proximity to different pathologies (e.g. plaques vs tangles vs vascular lesions).
Causality versus Correlation: It remains largely unknown which glial responses are truly pathogenic and which are protective or merely correlational. For instance, is microglial activation a primary driver of tau spread, or a failed attempt to contain it?
Inconclusive and Negative Findings: For many promising preclinical pathways (e.g. anti-inflammatories, TREM2 agonists), human clinical trial data have been inconclusive or negative. This suggests our preclinical models are poorly predictive, or that we misunderstand the therapeutic timing. Suppressing inflammation, for example, may be beneficial in mid-to-late stages but detrimental early on when microglial clearance functions are needed.
To address these gaps, we propose a focus on the research priorities outlined in Table 4.
Key knowledge gaps and future research priorities.

Future therapeutic strategies
Addressing the challenges of long-term glial modulation requires more refined strategies:
Intermittent Dosing Regimens: Rather than chronic, continuous drug delivery, intermittent dosing schedules (e.g. for CSF1R inhibitors) may allow glial cells to return to a basal homeostatic state, preserving beneficial functions while reducing inflammation 21 .
Pro-Resolving Mediators: Compounds such as resolvins, maresins, and lipoxins act not by blocking inflammation, but by actively promoting its resolution. These agents encourage glial cells to transition from a reactive to a reparative phenotype 18 .
Personalized, Biomarker-Driven Medicine: As patient-derived glial signatures vary, combining AI-powered analyses of transcriptomics with PET imaging and CSF biomarkers may help design tailored therapies that match individual neuroinflammatory states 22 .
In summary, future dementia treatments must embrace the complex, dynamic roles of glial cells. Rather than blunt-force suppression, fine-tuned modulation offers a sustainable path forward in achieving neuroprotection without undermining the essential support functions glial cells provide.
Conclusion
This review underscores a paradigm shift in dementia research, positioning glial cells not as passive bystanders but as active, dynamic regulators of neurodegeneration and neuroprotection. This review proposes a targeted, stage-specific glial modulation framework that balances therapeutic efficacy with the preservation of essential glial functions. By moving beyond traditional, neuron-centric models to account for glial heterogeneity, cross talk, and temporal evolution, we can better understand disease pathogenesis. With the rapid advancement of technologies like spatial transcriptomics, AI-integrated biomarker profiling, and human-relevant iPSC models, the vision of glia-informed, personalized dementia care is becoming increasingly attainable. Future studies grounded in this model can pave the way toward safer, more effective, and biologically attuned treatments that address the full complexity of brain aging and neurodegeneration.
Footnotes
Acknowledgements
None.
Ethical Considerations
Not applicable; this is a review paper.
Author Contributions
Hassan Jubair: Conceptualization, literature search, data extraction, analysis, writing (draft and editing), visualization, referencing, final approval.
Funding
The author received no financial support for the research, authorship, and/or publication of this article.
Declaration of Conflicting Interests
The author declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Data Availability Statement
Not Applicable.
Informed Consent/Patient Consent
Not Applicable.
Trial Registration Number/Date
Not Applicable.
Grant Number
Not Applicable.
Use of AI in Manuscript Preparation
The author utilized an AI language model (Gemini, Google) for language editing, grammar correction, and refinement of prose to improve clarity and flow. The author confirms that no scientific data, interpretations, or conclusions were generated or modified by AI.
AI-Generated Imagery
All figures in this manuscript were generated by the author using an AI image generation tool (OpenAI’s DALL·E 3 via ChatGPT). The author provided all conceptual input, scientific information, and labels, and iteratively refined the images to ensure scientific accuracy. These images are conceptual diagrams intended for illustrative purposes and do not represent original experimental data. OpenAI grants the author the right to use such generated content for academic and commercial purposes under its license agreement. Figures 1–5 were created by the author using OpenAI’s DALL·E 3 and conceptual diagrams, not original experimental data.
Statement of Human and Animal Rights
This article does not contain any studies with human or animal subjects.
Statement of Informed Consent
There are no human subjects in this article and informed consent is not applicable.