
Abstract
Cell-based therapies, particularly transplanted human oligodendrocyte progenitor cells (OPCs), are being explored for neuroprotection and remyelination in demyelinating diseases of the central nervous system (CNS). In this study, we investigated the potential of OPC transplantation into the optic nerve of dark agouti (DA) rats with experimental autoimmune encephalomyelitis (EAE). Human OPCs were transplanted 30 days after EAE induction in one optic nerve, while the contralateral nerve was injected with a vehicle. FTY720 (fingolimod) was administered starting from day 25 post-EAE to prevent graft rejection. Rats were monitored clinically and electrophysiologically using visually evoked potentials (VEPs) for up to 90 days post-transplant. Histological analysis of OPC viability, myelin, and axonal integrity was performed on days 30, 60, and 90 post-transplant. At days 30 and 60, sparse OPCs were detected in the injected optic nerve. However, no live cells were detected on day 90. There were no significant differences in myelin or axonal integrity between the OPC- and vehicle-injected nerves. The VEP traces were severely distorted throughout the 90-day follow-up. This approach did not show long-term viability following direct injection of OPCs in the optic nerve of EAE rats. Challenges related to graft rejection and cell transplantation are discussed, with implications for future research in cell-based therapies.

Keywords
Background
Failure of remyelination after repeated inflammatory damage leads to axonal degeneration and significant disability in patients with demyelinating diseases of the central nervous system (CNS), of which multiple sclerosis (MS) is the most common. MS is a chronic, immune-mediated disease in which autoreactive T and B cells target myelin antigens, triggering focal inflammatory demyelination, activation of microglia/macrophages, and secondary axonal degeneration that cumulatively drive irreversible neurological disability. While remyelination can occur spontaneously early in the disease, it gradually fails as oligodendrocyte progenitor cells (OPCs) become depleted or differentiation-incompetent, highlighting the need for regenerative strategies1,2. The currently available MS disease-modifying therapies mainly target the immune aspect and recurring inflammatory process characteristic of active demyelination. Although some of these treatments may slow down disability progression, they do not reverse the cumulative neurological damage caused by demyelination and subsequent axonal degeneration1,2. Cell-based therapies in demyelinating disease have generated substantial interest as novel therapeutic strategies for immune modulation, neuroprotection, or repair of damaged neurological tissue. Each approach has potential advantages, as well as safety concerns and unresolved questions 3 .
Oligodendrocytes (OLs) are glial cells responsible for myelin sheath formation in the CNS 4 . OLs originate from oligodendrocyte progenitor cells (OPCs), which are lineage-restricted, specialized cells with the ability to proliferate and differentiate5–7. The remyelination process in demyelinating disease is largely dependent on the recruitment of OPCs to the demyelinating lesion and the subsequent differentiation into new OLs. While OPCs are normally found within the adult CNS cell population, where they comprise 5%–8% of the total glial cells, their ability to maintain efficient remyelination in response to injuries is insufficient in chronic disease states8,9. Therefore, potential strategies that would restore this remyelination capability have become a major therapeutic target for preventing disease progression and accumulation of disability10,11. Previous studies have demonstrated that adult human OPCs can disperse through regions of experimental demyelination and are able to differentiate into myelinating OLs when injected into unaffected rat brains 12 . Therefore, it may be possible to inject the cells into selected locations and still obtain widespread repair 13 . In addition to their ability to differentiate into OLs, OPCs may play potential roles in immunomodulation, attenuation of the inflammatory process, and trophic support 7 .
The myelin-oligodendrocyte-glycoprotein (MOG₁₋₁₂₅)–induced experimental autoimmune encephalomyelitis (EAE) model in Dark Agouti (DA) rats closely mirrors key pathological and clinical features of relapsing–remitting MS, including immune-driven demyelination, and the occurrence of episodic neurological deficits. Previous studies have shown the inflammatory demyelination of the optic nerves in the DA rat following immunization with the (MOG)1-125 protein14,15. These rats typically develop a relapsing-remitting form of experimental autoimmune encephalomyelitis (EAE), in which the initial inflammatory attack is followed by demyelination 16 . We have previously described that demyelinating optic neuritis (ON) typically occurs around day 21–28 post-EAE induction and is correlated with electrophysiological evidence of demyelination on visually evoked potentials (VEP) 17 . Therefore, this animal model may be used for investigating the potential therapeutic benefits of OPCs.
In this study, we aimed to investigate the feasibility and therapeutic potential of locally transplanted human oligodendrocyte progenitor cells (OPCs) in promoting remyelination and preserving axonal integrity of the optic nerve in the DA-EAE rat model. Our second goal was to assess the utility of VEP recordings for monitoring the potential therapeutic benefit of OPCs injection.
Methods
Animals and EAE induction
EAE was induced in 20 Dark Agouti (DA) female rats aged 6–8 weeks with an initial body weight of 80–110 g. This sample size was chosen based on previous studies assessing optic nerve histology and VEP recordings in rats 18 . Animals were maintained in a controlled environment with a 12-h light/dark cycle and allowed unrestricted access to standard chow and tap water, following the guidelines set forth in the U.S. National Institutes of Health (NIH) Guide for the Care and Use of Laboratory Animals. All experimental protocols were reviewed and approved by the Animal Care and Use Committee (IACUC) at Massachusetts General Hospital (MGH).
For EAE induction, rats were anesthetized by inhaled isoflurane and injected subcutaneously with a total volume of 200 µl of inoculum containing 40 µg of rat recombinant MOG, corresponding to the N-terminal sequence of human MOG (amino acids 1–125; purchased from AnaSpec, Fremont, CA, USA), in complete Freund’s adjuvant (CFA), as previously described 18 .
OPCs transplantation
On day 30 post-EAE induction, rats were injected with 100,000 live OPCs derived from human induced pluripotent stem cells (iPSC) 19 into the left optic nerve, while the right optic nerve was injected with vehicle and served as control. Trypan blue exclusion was performed before injection to ensure cell viability of ≥ 90%. Optic nerve injections were performed using a minimally invasive technique to expose the optic nerve, as previously described 20 . In brief, the animal was shaved from the nose to the scruff of the neck, the skin was prepped with iodine and alcohol prep pads, and then the skin was opened in a midline linear fashion with gentle undermining to expose both upper lid fat pads. Sharp dissection revealed the orbital rim, and micro-rongeurs were used to remove the bony rim and expose the supra-orbital fat pad. Curved micro pickups were used to rotate the eye inferiorly, and the optic nerve was brought into view. The curved micro pickups were placed under the optic nerve to buttress during injection. The bolus of cells was delivered at a 1 µl/min rate through a glass-pulled pipette. We did not use growth factors in this study.
Immunosuppression protocol
Beginning from day 25 post-EAE induction (i.e. 5 days before OPCs transplantation), rats were given daily administration of FTY720 (fingolimod). FYT720 is a sphingosine 1-phosphate (S1P) modulator with known efficacy in suppressing disease activity in MOG-EAE in the DA rat21,22. Moreover, FTY720 has been shown to prevent graft rejection in rats with graft-versus-host disease and solid organ transplantation23–25. FTY720 was administered at approximately 0.5 mg/kg/day in the drinking water, as previously described 21 .
Clinical and electrophysiological assessment
Clinical EAE scores and VEPs recordings were carried out at baseline (i.e. before EAE induction) and on days 7, 14, 21, and 28 post-EAE induction. Following OPCs transplantation (i.e. day 30), EAE scores and VEPs recordings were performed at 2-week intervals during the first 30 days and at 4-week intervals during the following 60 days (i.e. days 45, 60, 90, and 120).
Clinical signs of EAE were evaluated on a scale from 1 to 5, based on established criteria: score 0, no observable disease; score 1, limp tail; score 2, limp tail and partial limb weakness: score 3, one hind limb paralyzed; score 4, both hind limbs paralyzed; score 5, moribund/dead 26 .
VEPs were recorded using a commercial system (Celleris, Diagnosys LLC, Lowell, MA, USA). Prior to each recording, rats were dark-adapted for 3–4 h in a dimly lit room. The recordings were conducted under anesthesia induced by ketamine (40 mg/kg) and xylazine (5 mg/kg). The body temperature was kept at 37°C, and pupil dilation was achieved with 1% tropicamide applied topically. We used the same flash VEP recording protocol as previously described (18). In brief, Flash VEP stimuli were presented at a frequency of 1 Hz and an intensity of 1 cd·s/m2. During each session, 50 white stimuli of 6500K color diamonds were delivered to each eye. These stimuli were used to measure the latency of the N1 wave within the P1-N1-P2 complex. The N1 latency for each session was determined as the average of the 50 measurements. Additionally, the amplitude of the N1 wave, defined as the peak-to-peak voltage between the preceding positive peak and the N1 peak, was calculated as the mean of 50 measurements taken during each recording session.
Histological assessment of OPCs viability
On days 30, 60, and 90 post-OPC transplantation (i.e. days 60–120 post-EAE induction), rats (n=7, 7, and 6, respectively) were sacrificed using Carbon dioxide (CO2) inhalation, and the optic nerves were harvested for histological assessment of OPCs viability. The optic nerves were fixed with 4% paraformaldehyde and subsequently embedded in paraffin. The tissue was analyzed using Luxol Fast Blue (LFB), immunohistochemistry, and immunofluorescent stainings. LFB Stain Kit (Cat. No ab150675, Abcam, MA, USA) was used per the manufacturer’s instructions. For the immunohistochemistry staining, deparaffinized specimens were incubated overnight at 4°C with primary antibodies against Olig2 (rabbit polyclonal, Cat. No PA5-85734, Thermo Fisher Scientific Inc., MA, USA; 1:500) and NEFH (mouse monoclonal, Cat No 13-1300, Thermo Fisher Scientific Inc., MA, USA; 1:20). ImmPRES Duet Double Staining Polymer Kit (Cat. No MP-7714-15, Vector Laboratories, CA, USA) was applied for visualization as the manufacture’s instruction.
After deparaffinization and antigen retrieval by citrate acid buffer (pH 6.0), primary antibodies against Human Nuclear Antigen (HuNu) (mouse monoclonal, Cat. No ab191181; Abcam, MA, USA; 1:200) and secondary antibodies (goat anti-mouse IgG(H+L) Alexa Fluor 488, Cat. No A32723; Invitrogen; CA, USA; 1:2000) were used to visualize injected human-origin OPCs. A Nikon C2 confocal microscope (Nikon, Tokyo, Japan) was used to acquire the images.
The area of demyelination and axonal integrity were quantified using the NIH-Elements AR Analysis software (version 5.21.03). The Automated Measurement function was utilized to automatically detect positive or negative lesions for LFB, Olig2, and NEFH staining. The percentage of demyelinating lesions was calculated based on the identified region of interest. All animals euthanized at each time point were included in the final analysis.
The study design is illustrated in Fig. 1.

Illustration of the study design.
This study has been reported in line with the ARRIVE guidelines 2.0.
Statistical analysis
Clinical EAE scores, as well as VEP latencies and amplitudes, are expressed as mean ± standard deviation (SD) or mean ± standard error of the mean (SEM). Group comparisons were conducted using the unpaired t-test with Welch’s correction. A P-value of less than 0.05 was considered statistically significant. All statistical analyses were performed using GraphPad Prism 9 for macOS (GraphPad Software, San Diego, CA, USA).
Results
EAE scores
No clinical EAE signs were observed during the first 13 days post-EAE. On days 14, 21, 28, and 30 post-EAE, the mean (SD) EAE scores were 0.25 (0.45), 0.53 (1.3), 1.6 (2.1), and 0.77 (1) respectively, reflecting a statistically significant increase compared to the baseline. The EAE score remained stable without EAE clinical signs throughout the post-OPC injection period (day 60–day 120) (Fig. 2).

Mean EAE scores were recorded at baseline and on days 7, 14, 21, 28, 30, 60, 90, and 120 post-EAE induction. Data are presented as the mean and standard error of the mean (SEM).
VEP latencies and amplitudes
The baseline mean (SD) N1 latency was 49.1 (2.3) milliseconds (msec). A mild prolongation in N1 latency was detected as early as day 7 post-EAE. On days 14, 21, 28, and 30 post-EAE, there was a significant prolongation of N1 latency compared to baseline, with the greatest delay observed on day 21 post-EAE (Fig. 3a). The baseline mean N1 amplitude was −7.4 µV ± 1.9 (median (range) = −7.5 (−12 to −3.3)). No significant changes in N1 amplitude were observed on days 14, 21, 28, or 30 post-EAE induction (Fig. 3b). The VEP traces recorded after OPC injection (days 60–120 post-EAE induction) were severely distorted. Therefore, we were not able to calculate the N1 latencies and amplitudes during this period of the study.

Mean N1 VEP latencies (A, in milliseconds) and amplitudes (B, in µV) were recorded at baseline and on days 7, 14, 21 28, and 30 post-EAE induction. Data are presented as the mean and standard error of the mean (SEM). Due to the severe distortion of the VEP recordings, it was not possible to calculate the N1 latencies and amplitudes in the post-OPC period (i.e. days 60–120).
Demyelination and axonal preservation
No significant difference in the extent of demyelination and axonal preservation between OPC-injected and vehicle-injected optic nerves was observed throughout the study (Figs 4 and 5). Histology staining on day 30 post-OPC injection showed partial demyelination in both the OPC- injected and vehicle-injected optic nerves, with no statistically significant difference between the two groups (mean % of demyelination in OPC-injected optic nerves = 22.8 ± 9.1; mean % of demyelination in vehicle-injected optic nerves= 39.6 ± 22.5; P = 0.48). On day 60, the mean % of demyelination in the OPC-injected optic nerves was 18.62 ± 22.1, while the mean % of demyelination in vehicle-injected optic nerves was 10.1 ± 4.4 (P > 0.99). On day 90, the mean % of demyelination in the OPC-injected optic nerves was 47.36 ± 22.7, and the mean % of demyelination in vehicle-injected optic nerves was 88.24 ± 16.6 (P = 0.33).

Optic nerve sections 30 (a, b) and 60 (c) days after injection of human OPCs were treated with anti-HuNu antibody for immunofluorescence staining. At 30 days after injection, anti-HuNu antibody-positive cells with smaller nuclei than rat astrocyte/oligodendrocyte cluster in the OPC-injected area (a, center left), and some cells are scattered in the tissue (a and b, red arrows). A few anti-HuNu antibody-positive cells per slide can still be seen 60 days after injection (c, red arrow) but not in the optic nerve 90 days after injection. Blue: DAPI; green: anti-HuNu antibody.
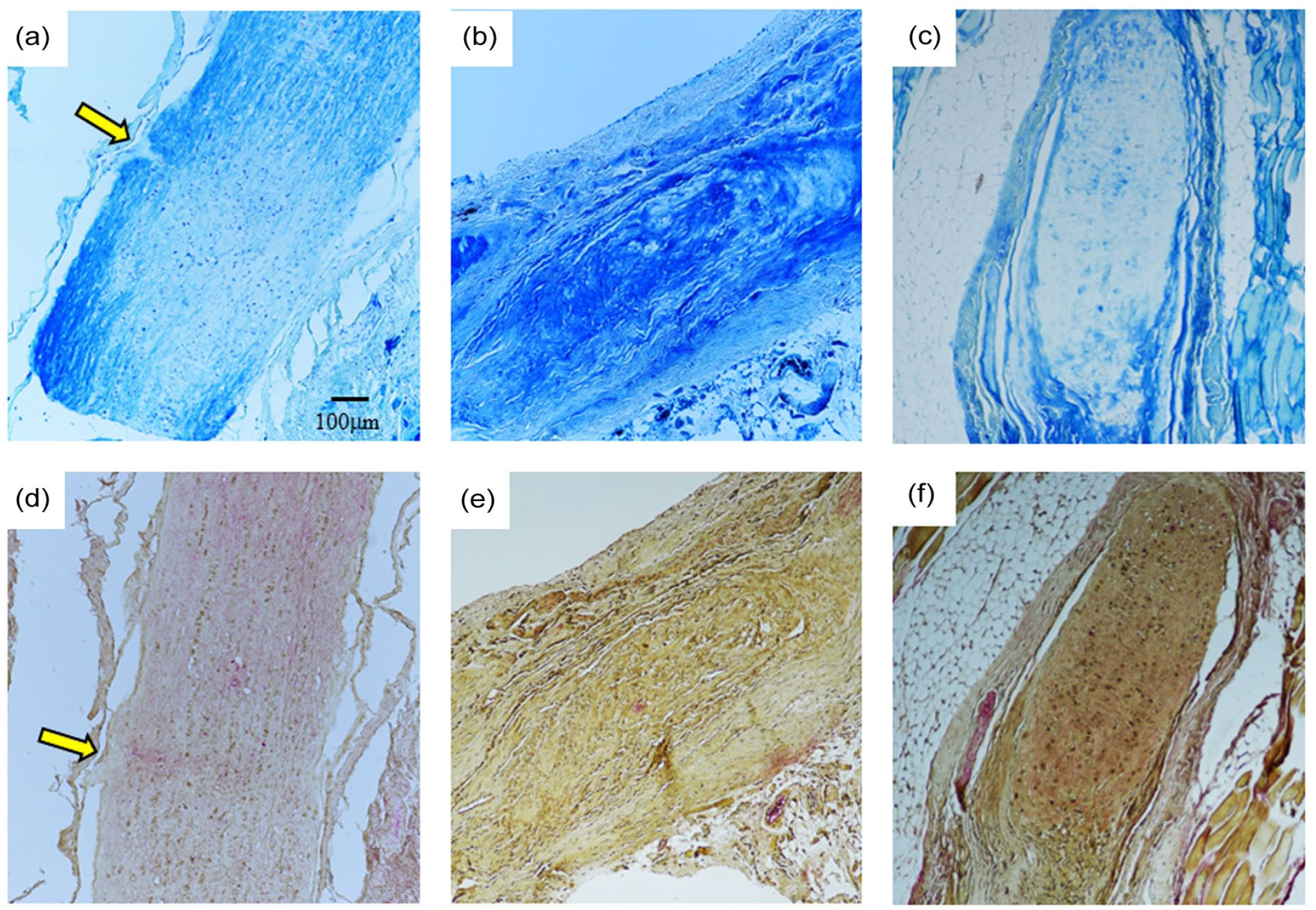
LFB and Olig2/NEFH double staining of the optic nerve after OPC injection. Yellow arrows indicate the injection site. On day 30, the optic nerve at the injection site shows partial loss of LFB staining (a). The same area shows a slight increase in NEFH staining by Olig2/NEFH double staining but no change in Olig2 (d). Scattered areas of decreased LFB staining are seen after 60 days (b). Olig2 staining is relatively preserved, with patchy-elevated NEFH staining (e). On day 90, the optic nerve shows decreased LFB staining areas (c), while Olig2 stainability was preserved (f).
The mean % of Olig2 staining on day 30 in the OPC-injected optic nerves was 28.9 ± 18.8 versus 33.17 ± 44.8 in the vehicle-injected optic nerves (P = 0.88). On day 60, The mean % of Olig2 staining in the OPC-injected optic nerves was 39.2 ± 15.6, and the mean % of Olig2 staining in the vehicle- injected optic nerves was 9.6 ± 1.9 (P = 0.1). The mean % of Olig2 staining on day 90 in the OPC- injected and vehicle-injected optic nerves was 54.93 ± 35.6 and 20.38 ± 5.4, respectively (P = 0.33). The mean % of NEFH staining in the OPC-injected and vehicle-injected optic nerves on days 30, 60, and 90 was 1.89 ± 2.2 versus 0 (P = 0.43), 2 ± 2.1 versus 0.63 ± 1.1 (P = 0.7), and 7.54 ± 10.5 versus 3.66 ± 1.5 (P > 0.99), respectively.
OPCs viability
Representative images of HuNu staining for the detection of OPCs on days 30, 60, and 90 post-OPC injection into the optic nerve are shown in Fig. 6. On days 30 and 60 post-OPC injection, a few sparse cells were detected in the injected tissue. However, on day 90 post-OPC injection, we were unable to detect live cells in the optic nerve. A population analysis of DAPI-positive cells and HuNu-positive cells demonstrated 16.01% ± 15.13% of HuNU-positive cells on day 30, and 8.15% ± 6.7% of HuNu-positive cells on day 60.

LFB staining and Olig2/NEFH immunohistochemical double-stained pathology images were used for quantitative analysis of the area of demyelination and axonal preservation. Data are presented as the mean and standard error of the mean (SEM). All comparisons between OPC-injected and vehicle-injected optic nerves are not statistically significant.
Discussion
Promoting regeneration to restore CNS function and prevent disability accumulation represents a major therapeutic challenge in chronic demyelinating diseases. OPCs play a crucial role in the physiologic regenerative response to demyelination and are considered a promising therapeutic target to promote remyelination in demyelinating diseases27,28. The majority of remyelinating strategies focus on the direct transplantation of OPCs or OLs or on targeting various intrinsic signaling mechanisms in the differentiation process of OLs 28 . In the current study, we describe our experience with direct transplantation of OPCs into the optic nerve in the MOG 1-125–EAE DA rat model.
The direct transplantation of OPCs into demyelinating lesions in rodents poses several technical and safety challenges. Because injection of human OPCs in rodents implies the use of allogeneic cells, immunosuppression of the recipient animal is required to prevent rejection, especially in a xenografting setting 3 . However, there is insufficient data regarding the best protocol for immunosuppression in this context 29 . We used FTY720 (fingolimod), an S1P modulator that was administered in the drinking water. Our choice of using FTY720 was based on its known efficacy in reducing EAE disease activity21,22, as well as its efficacy in preventing graft rejection in rats with graft-versus-host disease and solid organ transplantation23–25. While our rats did not have further EAE attacks while on the medication, the fact that we could not demonstrate viable OPCs after 60 days in the injected optic nerves may suggest it may not be as effective in preventing rejection of human OPCs, and additional immunosuppressants, such as Cyclosporin A 23 will need to be tested. The administration of FTY720 in the drinking water warrants consideration as well. Although this method has been previously reported to be effective in ameliorating the clinical scores in EAE 21 , it may be prone to more variability in the drug concentrations compared to intravenous or subcutaneous administration, making it less appropriate for the prevention of graft rejection.
Another challenge of OPC transplantation in rodents is the technical difficulty of injecting the cells into a small-caliber nerve. In fact, the rodent’s optic nerve diameter is approximately 1 mm 30 , making it difficult for the nerve to accept a volume of the cell bolus. This may explain the lack of success in cell-replacement and restoration therapeutic approaches attempted in small-caliber nerves (e.g. the sciatic nerve, which has a diameter comparable to the optic nerve).
An important observation of our study is that the VEP recordings after OPCs injection were severely distorted for up to 90 days after the injection procedure, even when saline alone was injected, making the electrophysiological evaluation of the optic nerve after injections not feasible. We believe that the alteration of the VEPs recording likely reflects permanent damage caused by local trauma during the injection procedure. Therefore, using VEPs to evaluate regenerative therapies may be more appropriate in therapeutic approaches that do not involve local trauma to the optic nerve.
In addition to their role in remyelination, OPCs have been increasingly recognized for their immunomodulatory properties, including the ability to promote the secretion of anti-inflammatory cytokines, modulate microglial activation, and influence the balance of T cell subsets in inflammatory CNS environments 31 . These dual regenerative and immunological functions may operate in parallel or interact synergistically. Although our study was not designed to directly assess immune modulation, future studies incorporating cytokine profiling and immune cell phenotyping may help elucidate how these dual roles contribute to tissue repair.
Several limitations of our study should be considered. First, we did not characterize the inflammatory microenvironment at the time of OPCs transplantation. Iba1 staining for microglial activation and markers of astrocytosis or macrophage infiltration may have helped clarify whether local inflammation contributed to the observed loss of OPC viability or the absence of remyelination. Future studies should incorporate comprehensive immunohistochemical profiling to better characterize the transplantation niche within the optic nerve and its permissiveness for regeneration. Such an approach would also enable to optimize the timing of OPCs transplantation. Second, co-staining with OLIG2 and HuNu to confirm the oligodendrocyte lineage of the transplanted cells was not performed. Third, we did not assess in vivo differentiation of the transplanted OPCs into mature oligodendrocytes. Future studies should incorporate markers of differentiation such as MBP, CNPase, or O1 to determine whether the transplanted OPCs are able to differentiate into mature myelinating cells.
Conclusion
Our preliminary study of OPCs injection into the rat’s optic nerve following EAE-ON could not confirm the long-term viability of human OPCs in our model. Additionally, the VEP recordings after OPCs injection were severely distorted during the 90-day post-injection follow-up period, making it impossible to evaluate the potential electrophysiological regenerative properties of this approach. Future studies focusing on cell therapy in ON should consider an improved immunosuppressive treatment to mitigate xenogeneic graft rejection or alternative strategies, like intravenous or intrathecal administration of exogenous cells that could potentially migrate and modulate the host immune system, generating an anti-inflammatory, pro-regenerative environment. Additional characterization of the immune environment and cell differentiation is also warranted.
Footnotes
Acknowledgements
Not applicable.
Ethical considerations
All the procedures described in this manuscript were approved by the Massachusetts General Hospital (MGH) Animal Care and Use Committee (IACUC). Title of approved project: Regenerative Neural Stem Cell Transplantation in Rat Model of Demyelinating Disease; Name of the institutional approval committee: Massachusetts General Hospital (MGH) Animal Care and Use Committee (IACUC); Approval number: 2019N000180; Date of approval: 1 November 2019.
Consent to participate
Not applicable.
Consent for publication
Not applicable.
Author contributions
Itay Lotan: Conception/design, Data Acquisition, Analysis and Interpretation, Drafting and revising the manuscript.
Benjamin R Johnston: Data Acquisition, Analysis and Interpretation, Drafting the manuscript.
Shuhei Nishiyama: Data Acquisition, Analysis and Interpretation, Drafting the manuscript.
Jin Myoung Seok: Data Acquisition, Analysis and Interpretation, Drafting the manuscript.
Amy Wright: Data Acquisition, Analysis and Interpretation, Drafting the manuscript.
Stanley Bazarek: Data Acquisition, Analysis and Interpretation, Drafting the manuscript.
Michael Levy: Conception/design, Analysis and Interpretation, Supervision, Drafting and revising the manuscript.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was supported by BlueRock Therapeutics (Cambridge, MA, USA).
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Data availability statement
The datasets used and/or analyzed during the current study are available from the corresponding author upon reasonable request.
Statement of human and animal rights
This article does not contain any studies with human or animal subjects.
Statement of informed consent
There are no human subjects in this article and informed consent is not applicable.