
Abstract
Systemic lupus erythematosus (SLE) is a chronic autoimmune disorder characterized by widespread immune dysregulation and systemic inflammation. Among its many manifestations, lupus nephritis (LN) stands out as one of the most severe and life-threatening complications, affecting up to 60% of SLE patients. LN primarily results from a type III hypersensitivity reaction in which immune complexes are deposited within the kidney. This leads to progressive glomerular and tubulointerstitial lesions compromising renal function. A significant portion of patients presenting with LN will progress to end-stage renal disease (ERSD). Despite advances in treatment strategies, including standard and targeted biologic immunosuppressants, many patients with LN fail to achieve long-term remission, leaving a significant need for safer and more effective therapies. In recent years, T-cell-based therapies have emerged as a promising frontier in the treatment of a variety of conditions including autoimmune diseases. Specifically, chimeric antigen receptor (CAR) T-cell therapies aimed at depleting antibody-producing B cells have demonstrated the ability to restore immune tolerance in several preclinical models of diseases with B-cell-driven pathologies, including SLE. In the case of LN, CAR T-cell therapies have also been deployed in clinical trials to treat patients with refractory disease. The positive results of initial clinical trials provide strong evidence that B-cell-targeted cellular therapies such as CAR T cells targeting B-cell markers CD19 and B-cell maturation antigen (BCMA) might be an effective modality for rebalancing the immune system through the elimination of autoreactive B cells. This review examines the current state of CAR T-cell therapies and their applications in LN by exploring the mechanisms, challenges, and the potential of this unique treatment approach.

Introduction
Systemic lupus erythematosus (SLE) is a chronic autoimmune disease characterized by widespread inflammation and damage to various organ systems in the body 1 . Lupus nephritis (LN) refers to the clinical manifestation of SLE within the kidneys. This represents one of the most clinically significant aspects of the disease. Approximately 50% of adults with SLE will develop LN, and nearly 80% of pediatric cases manifest in LN. Moreover, epidemiological studies reveal a higher incidence of LN in non-white women, indicating potential genetic and environmental influences on disease susceptibility2,3.
The pathophysiology of LN involves a complex interaction between the immune system and kidney tissue. In LN, the immune system erroneously targets the kidneys, leading to inflammation and injury 4 . This aberrant immune response is primarily driven by the production of autoantibodies, particularly anti-double-stranded DNA antibodies, which manifest in a type III hypersensitivity reaction involving deposition of immune complexes in the kidney’s glomeruli 5 . These immune complexes can activate the complement system, triggering a cascade of inflammatory events characterized by cytokine release and the recruitment of immune cells into the renal tissue 6 . This inflammatory process results in significant damage to both the glomerular and tubulointerstitial regions of the kidneys, impairing their ability to function 7 .
Clinically, LN manifests with a wide range of symptoms and severity. Some patients present with abnormal lab tests and few other symptoms while patients with more severe cases present with aspects of nephritic and/or nephrotic syndromes such as proteinuria, hematuria, acute renal failure, or hypertension, often indicating more progressive renal dysfunction 8 . If left untreated, it can progress to end-stage renal disease (ESRD), necessitating dialysis or kidney transplantation 9 . The variability in clinical presentation and histological findings in LN underscores its complex pathogenesis, influenced by a combination of genetic predisposition, environmental triggers, and dysregulated immune responses. Currently, the treatment of LN relies heavily on immunosuppressive and anti-inflammatory therapies aimed at controlling disease activity and preventing renal damage. Standard treatments include high-dose corticosteroids, alkylating agents like cyclophosphamide, or antimetabolites such as mycophenolate mofetil (MMF). In addition, biological therapies, including B-cell-depleting monoclonal antibodies like belimumab, have gained attention in recent years 10 . Despite these advancements, many patients experience inadequate responses, relapses, and are subjected to potentially severe treatment-related side effects, including infections, organ toxicity, and carcinogenesis. This highlights an urgent need for novel therapeutic approaches that are both effective and safer for long-term disease management. Emerging therapies, particularly T-cell-based strategies, are being explored for their potential to address the underlying immunopathology of LN while minimizing off-target effects and improving patient outcomes.
LN pathology
LN is a prototypical immune complex-mediated glomerulonephritis, where complement and antibodies are deposited in the glomeruli, causing local inflammation. The pathogenesis of LN involves a complex interplay of both innate and adaptive immune cells, particularly B and T cells, which are central to disease progression11,12.
The pivotal roles played by B cells in the pathogenesis of LN stem from their ability to produce autoantibodies, present antigens, and secrete cytokines. In SLE, B cells lose tolerance to self-antigens, leading to the production of autoantibodies against nuclear components such as double-stranded DNA (dsDNA). These autoantibodies form immune complexes that deposit in the kidneys, triggering inflammation and renal tissue hyperplasia, resulting in abnormal renal function13,14. B cells in LN exhibit heightened activation, and immune memory is fixed by progressive differentiation into plasma cells which are responsible for sustained autoantibody production. Notably, long-lived plasma cells residing in the bone marrow continue to produce autoantibodies independent of ongoing B-cell activation, driving chronic inflammation and progressive kidney damage 15 .
T cells also play a critical role in the pathogenesis of LN. The role of CD4+ T helper cells, particularly Th1 and Th17 subsets, involves production of type I interferons (IFNs) that promote activation of autoreactive B cells. Activated CD4+ T cells are also involved in the production of proinflammatory cytokines16–18 which enhance the recruitment and activation of innate immune cells, including macrophages and neutrophils, to the kidneys18–20. Follicular helper T cells are involved in somatic hypermutation of B-cell receptor genes which drives the manifestation of high-affinity autoantibodies. While T cells clearly play a role in LN, they primarily drive disease progression through effector functions leading to persistent innate and humoral immune activation21,22.
Histologically, LN can be classified into five distinct classes based on the World Health Organization classification, each reflecting different patterns of renal injury, ranging from minimal change disease to diffuse proliferative glomerulonephritis12,23. The mechanisms of kidney damage in LN are multifactorial and involve several key processes. The deposition of immune complexes in the glomeruli initiates an inflammatory cascade, activating complement pathways and recruiting inflammatory cells to the renal tissue24,25. B-cell-derived autoantibodies can activate the complement system, which plays a dual role in LN5,26,27. While classical complement activation can aid in clearing immune complexes, chronic activation, particularly of the alternative pathway, can exacerbate tissue damage through the formation of the membrane attack complex and the release of anaphylatoxins like C3a and C5a, which escalate inflammation and promote further immune cell recruitment5,26,27. Proinflammatory cytokines such as tumor necrosis factor alpha (TNF-α), interleukin 6 (IL-6), and type I IFNs released by various immune cells contribute to the kidney’s inflammatory environment, enhancing B-cell activation and promoting T-cell proliferation 28 . In addition, chronic inflammation leads to hyperplasia of endothelial cells, mesangial cells, and podocytes within the kidney. Furthermore, these cells along with activated myofibroblasts produce excessive extracellular matrix resulting in progressive renal fibrosis29–31. This basic model of disease-causing pathways in LN, as depicted in Fig. 1, has led to the identification of potential therapeutic targets designed to modulate the immune response, reducing inflammation, and protect against kidney damage in patients with LN.

A schematic depicting the pathogenesis of lupus nephritis (LN) and its effects on the kidney and liver. In systemic lupus erythematosus (SLE), loss of B-cell tolerance leads to autoantibody production against nuclear components, such as double-stranded DNA (dsDNA). Autoreactive B cells differentiate into long-lived plasma cells, sustaining chronic autoantibody production. CD4+ T helper cells, particularly Th1 and Th17 subsets, enhance B-cell activation and inflammatory cytokine production, recruiting immune cells, such as neutrophils and macrophages, to the kidneys. Autoantibody production leads to immune complex formation and deposition in the kidneys, triggering inflammation and activating the alternative complement pathway. Chronic inflammation results in hyperplasia of endothelial, mesangial, and podocyte cells, along with excessive extracellular matrix deposition, leading to kidney damage and, in some cases, progressive fibrosis. In addition, systemic inflammation and immune complex deposition can contribute to renal dysfunction, further exacerbating disease pathology.
Current therapies for LN
Current therapies for LN primarily focus on suppressing immune activation, preserving kidney function, and preventing disease progression. Therapeutic strategies encompass a combination of immunosuppressive agents, biologics, and supportive care tailored to the severity of the disease and patient-specific factors32,33.
Broad-spectrum immunosuppressants remain the cornerstone of treatment, with corticosteroids like prednisone and methylprednisolone frequently used to rapidly reduce inflammation during disease flares 34 . MMF is a first-line therapy for both induction and maintenance phases of treatment due to its ability to inhibit lymphocyte proliferation35,36, while cyclophosphamide, an alkylating agent, is reserved for severe cases, particularly in patients with proliferative glomerulonephritis 37 because of increased toxicity and negative impact on fertility 38 . Azathioprine is often employed as maintenance therapy, especially in those unable to tolerate MMF35,39. However, despite cases where these treatments are efficacious, these agents can have significant side effects, prompting the search for more targeted approaches that lead to more durable remissions 34 .
Biologic therapies have transformed the treatment landscape of most autoimmune disease by offering precision in targeting the immune pathways underlying the disease state. In LN, rituximab, a monoclonal antibody targeting CD20 on B cells, depletes autoreactive B cells and is still used in refractory cases of LN despite not meeting its primary endpoints in clinical trials40–42. A newer anti-CD20 biologic, obinutuzumab (GA101), demonstrated positive phase III results and may be more effective than standard therapy alone in depleting pathogenic B cells in LN when used in combination with standard therapy. Notably, patients receiving obinutuzumab achieved a higher percentage of complete renal response 43 . Belimumab, the first Food and Drugs Administration–approved biologic for LN, inhibits B-cell activating factor (BAFF), reducing B-cell survival and autoantibody production while lowering disease activity and flares 44 . Similarly, anifrolumab targets the type I IFN receptor, dampening the IFN-driven inflammatory cascade that contributes to LN pathogenesis, and is currently being studied in LN patients 45 .
Complement targeted therapies are also being explored for their role in mitigating immune complex-mediated kidney damage. Eculizumab, a monoclonal antibody against C5, prevents formation of the membrane attach complex (MAC) and has shown promise in refractory LN 46 . Ravulizumab, which targets the leukocyte integrin MAC-1 (CD11b/CD18), or complement receptor (CR3), was investigated in the Sanctuary trial for indications including IgA neuropathy and LN although specific outcomes for the LN cohort have yet to be disclosed 47 . Other agents targeting upstream components of the complement cascade, such as avacopan, a C5a receptor inhibitor 48 , and newer C3 inhibitors such as pegcetacoplan 49 and compstatin derivatives 50 , are under active investigation. These therapies aim to interrupt the amplification loop of inflammation driven by complement activation, offering another targeted approach that could improve renal outcomes in LN.
Supportive therapies, including hydroxychloroquine (HCQ) to reduce disease flares 51 and renin-angiotensin-aldosterone system (RAAS) blockers to manage proteinuria and hypertension52,53, are critical adjuncts to immunosuppressive regimens. In addition, lifestyle modifications to improve blood pressure control with routine monitoring of kidney function are essential for optimizing long-term outcomes. Despite the progress in LN treatment, these therapies still rely on broad immunosuppression, often resulting in significant toxicity and incomplete disease control54,55. This underscores the need for novel approaches that target alternative immune pathways, including innovative approaches like adoptive cell transfer designed to promote broad tolerance to self-antigens.
Adoptively transferred cellular therapies for LN
Adoptive transfer of autologous hematopoietic stem cells (HSCs) emerged as a tool in rheumatic disease when studies in lupus-prone mice demonstrated its potential to prevent the onset of autoimmune disease. Clinically, hematopoietic stem cell transplantation (HSCT) has been studied and utilized in certain refractory cases, particularly when co-indicated for a malignant disease. However, due to the significant complications and risk of mortality associated with HSCT, it is considered a last-line therapy, and its use in LN has declined over the past decade 56 .
In pursuit of safer and more targeted alternatives, other experimental therapies involving the adoptive transfer of mesenchymal stromal cells (MSCs) and T regulatory cells (Tregs) have yielded promising results in preclinical models. Neither modality has yet demonstrated significant clinical remission, but a phase II trial for efficacy of MSCs is currently underway. The rationale behind both strategies is to induce remission by a variety of proposed mechanisms, which have been reviewed in detail elsewhere 57 .
With the emergence of advanced cellular engineering technologies, new immunotherapeutic approaches are being explored as potential alternatives. Among these, engineered lymphocyte-based approaches have shown promise in preclinical studies and are currently being evaluated in clinical trials. In the next section, we discuss how engineered lymphocytes that are redirected to target pathogenic B cells are emerging as promising tools for achieving durable remissions in patients with rheumatic diseases, including LN.
Engineered T cells as therapeutic agents for LN
Engineered T-cell therapies are based on the principle of redirecting cytotoxic T-cell specificity by modifying them to express engineered receptors that bind to predefined targets. Chimeric antigen receptor (CAR) molecules are generated by fusing intracellular T-cell-activating motifs (CD3z, 41BB, CD28, etc.) with an extracellular motif containing a single-chain variable fragment (scFv) that specifically binds to a target molecule on the surface of the intended cell as depicted in Fig. 2. T cells engineered to express these CAR molecules are referred to as CAR T cells 58 . CAR T-cell therapies have been most successfully deployed for the treatment of hematological malignancies which express the pan-B-cell marker CD1959,60. An expected side effect of this kind of therapy is B-cell aplasia, which typically persists for several months following treatment, but ultimately, most patients recover B-cell counts within a year as the number of adoptively transferred T cells decline61–63. This potent, transient B-cell-depletion phenomenon serves as the basis of using CD19-redirected CAR T cells for the treatment of LN. Indeed, preclinical studies have provided evidence that CD19 CAR T-cell–induced B-cell aplasia is effective in mouse models of lupus64,65.
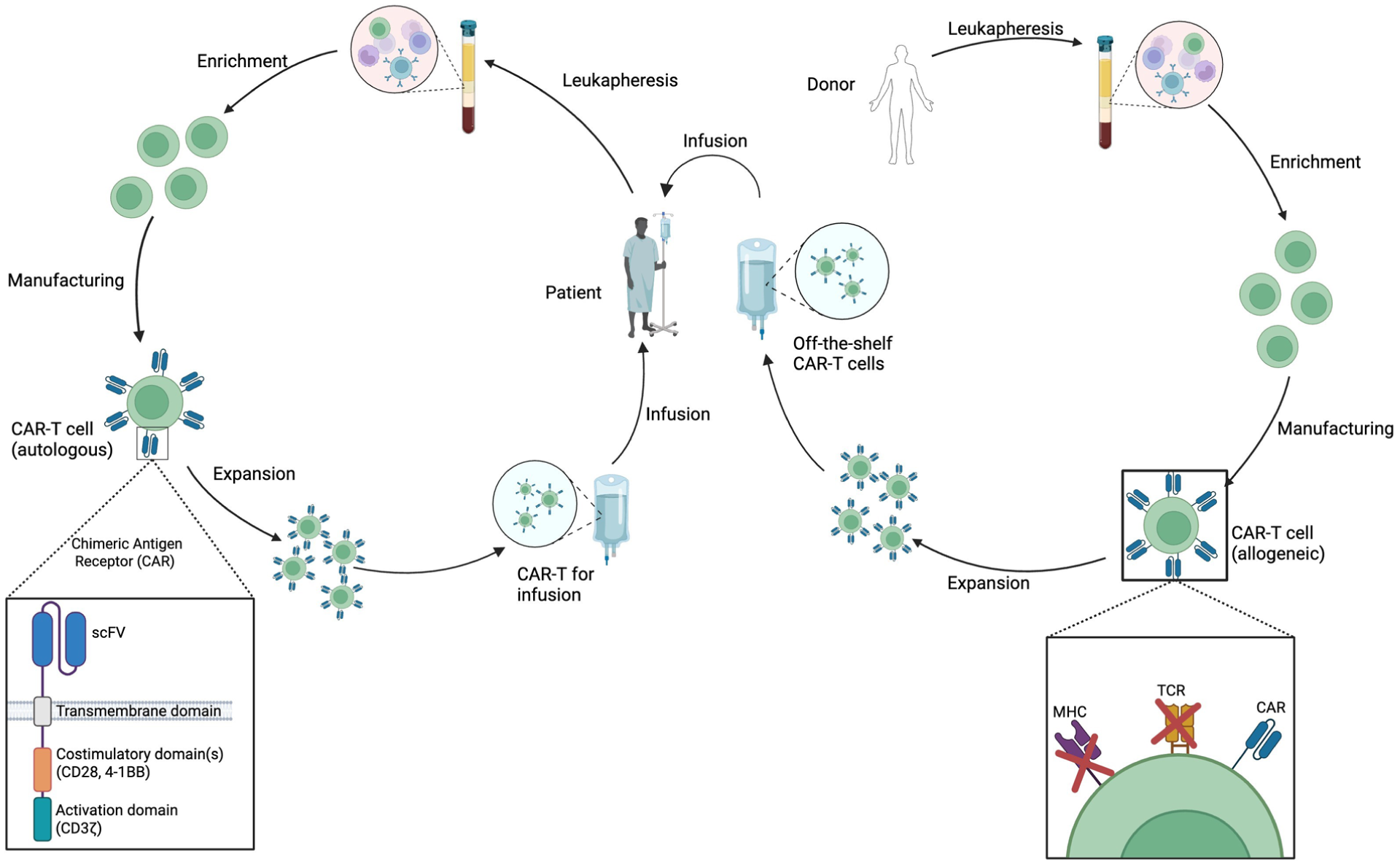
Schematic illustrating autologous versus allogeneic CAR T-cell manufacturing. Left: In autologous CAR T-cell therapy, T lymphocytes are isolated from a patient’s peripheral blood mononuclear cells (PBMCs). Chimeric antigen receptors (CARs) are introduced via genetic engineering methods (e.g., viral transduction or transfection), and the modified T cells are expanded before being reinfused into the same patient. Right: In allogeneic CAR-T therapy, T lymphocytes from a healthy donor are engineered to express CARs while simultaneously disrupting endogenous T-cell receptors (TCR) and major histocompatibility complex (MHC) molecules to reduce the risk of graft-versus-host disease (GvHD) and host rejection. The modified cells are expanded, purified, and cryopreserved, creating an “off-the-shelf” CAR T-cell product suitable for use across multiple patients.
In addition to CD19, B-cell maturation antigen (BCMA) has emerged as another promising target for CAR T-cell therapy. While BCMA has primarily been studied in the context of multiple myeloma66,67, a malignancy resulting from plasmablast-like cells expressing BCMA, it is also expressed on mature B cells, plasmablasts, and plasma cells. CAR-T therapies targeting BCMA have shown the ability to rapidly deplete these cell types in patients. Taken together, these observations suggest that CAR-T therapy might offer an opportunity to address limitations in current treatments by more effectively targeting a critical component of immune dysregulation in LN68,69.
Another novel T-cell-based therapeutic strategy involves the use of ex vivo expanded Epstein-Barr virus (EBV)-specific T cells. Latent EBV infection can lead to long-term persistence of infected B cells, which can exhibit a hyperactivated phenotype and act as a persistent source of immune stimulation, which have been implicated in the pathogenesis of SLE and LN70–72. This is the rational for studies that have examined the utility of adoptive transfer of EBV-targeted T cells as a way to selectively eliminate these infected, autoreactive B cells. Early-phase studies have shown that autologous EBV-specific T-cell therapy is well-tolerated; however, it is not clear if it may lead to reductions in disease activity 73 .
The development of chimeric autoantibody receptor (CAAR) T cells represents another innovative strategy aimed at eliminating autoreactive B cells with high specificity 74 . CAAR T cells are engineered to express autoantigenic epitopes on their extracellular domain, allowing them to selectively bind and kill B cells that produce pathogenic autoantibodies. In contrast to traditional CAR T cells, which recognize surface proteins like CD19 or BCMA, CAAR T cells mimic the antigen itself, targeting only the B cells responsible for producing disease-specific antibodies 75 . This approach has been explored in preclinical models of autoimmune diseases such as pemphigus vulgaris 75 and myasthenia gravis 76 , where CAAR T cells depleted autoreactive B cells while sparing healthy immune cells. More recently, this strategy has recently been extended to LN, where CAAR T cells directed against anti-dsDNA producing B cells demonstrated enhanced selective cytotoxicity in vitro 77 . These early findings suggest that CAAR T-cell therapy could represent a safer, more targeted alternative to broad immunosuppression in LN, with the potential to achieve durable disease control through the elimination of pathogenic B-cell subsets.
Unlike conventional immunosuppressive agents, T-cell-based therapies have the advantage of selectively targeting specific pathogenic cellular subsets. This targeted approach reduces off-target effects and lowers the risks associated with broad immune suppression, such as increased risk of infections and malignancies78,79. In LN, where B-cell activation is a central driver of disease, CAR-T cells targeting B-cell markers, such as CD19 and BCMA, have shown promise in reducing autoantibody production and disrupting the autoimmune cascade80,81. By depleting autoreactive B cells, these CAR T cells address a critical aspect of LN pathogenesis. In addition, these therapies have the potential to restore immune tolerance, addressing the root cause of autoimmunity rather than merely suppressing symptoms68,82.
Several ongoing clinical trials are investigating the use of CD19, CD20, and BCMA CAR T-cell therapies for LN80,81. These trials aim to evaluate the safety and efficacy of these innovative therapies, with specific objectives including the reduction of disease activity, improvement in renal function, and decreased autoantibody production. Preliminary results from these trials are promising, indicating potential efficacy in managing refractory or severe LN and highlighting the need for further investigation to confirm these findings.
Emerging research
Emerging research using animal models of lupus offers exciting new insights into the potential of targeted T-cell therapies. These studies have demonstrated that depleting specific B-cell populations can reduce disease severity and renal damage. For instance, several preclinical studies have been published investigating the utility of CD19 CAR T cells for depleting B cells in murine models of lupus. These studies leveraged retroviral transduction of mouse T cells with a CAR construct targeting mouse CD19 (scFv clone ID3) and observed extended B-cell aplasia in several treated animals64,65. While promising, preclinical efforts to target alternative B-cell antigens, like BCMA and CD20, have been limited by the unavailability of suitable murine BCMA and CD20 CAR constructs. However, the success reported in these studies has led to several clinical trials investigating CAR T cells targeting various B-cell antigens including CD19, CD20, and BCMA in autoimmune disease, such as lupus (Table 1). These studies are based on the presumed expression of these antigens on lupus-associated B cells and autoreactive plasmablasts, ultimately supporting the potential of CAR T-cell therapies as a novel therapeutic strategy to target the autoimmune dysregulation driving lupus68,83–86.
Ongoing and completed clinical trials investigating CAR T-cell therapies targeting B-cell antigens in autoimmune diseases.
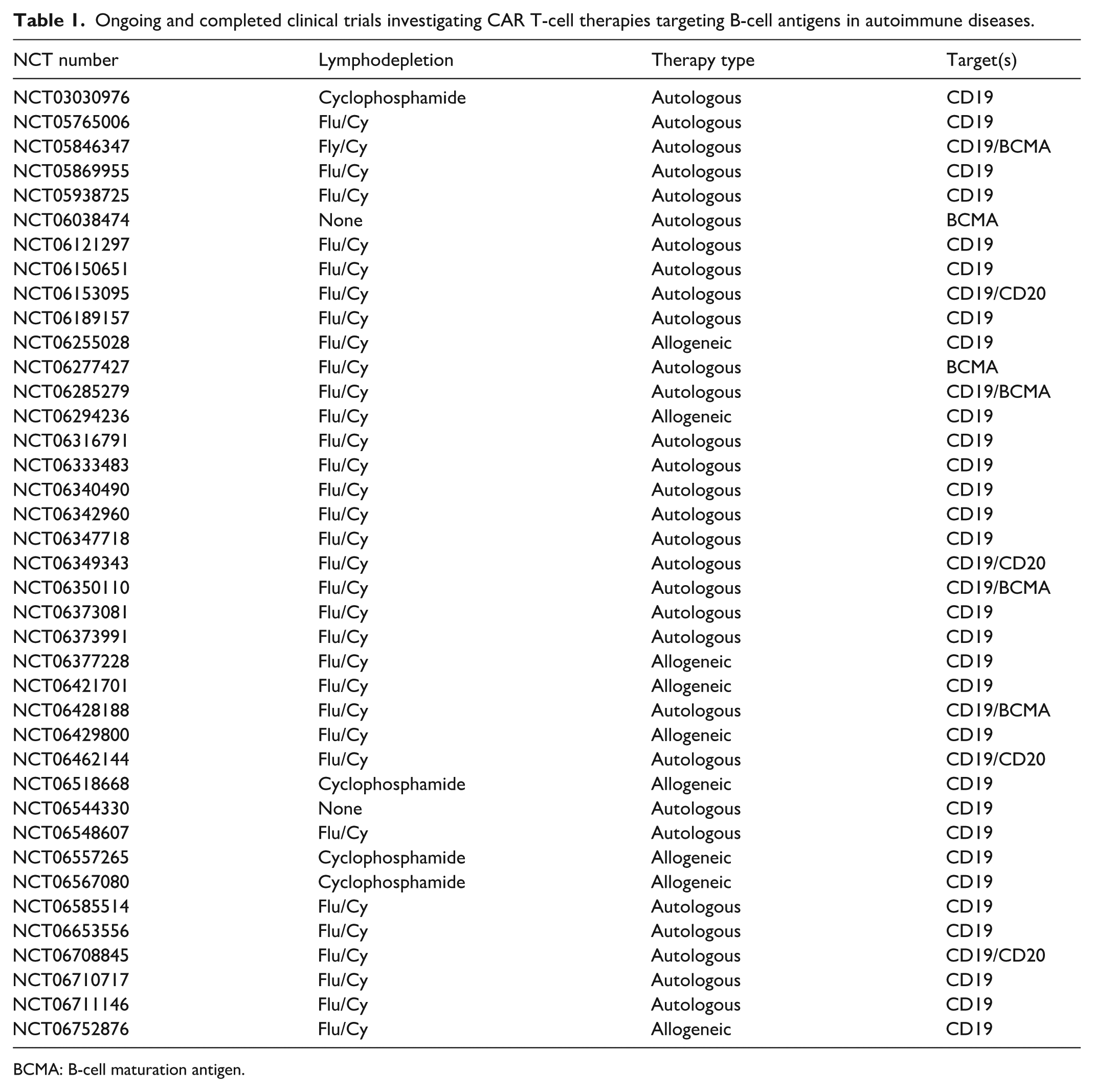
BCMA: B-cell maturation antigen.
In parallel with these animal studies, other research aims to address the aforementioned challenges associated with allogeneic CAR-T-based therapies in LN. Some are based on engineered conventional T cells that have had the T-cell receptor locus knocked out to eliminate graft-versus-host disease (GvHD), a major complication that must be addressed in any off-the-shelf T-cell therapy87,88. Alternative methods involve engineering natural killer T cells (NK-T cells) 89 , gamma-delta T cells (γδ T cells) 90 , or Tregs91,92 which inherently do not cause GvHD. Similarly, RNA-based engineering strategies are being developed to utilize RNA encoding a CAR that is introduced into T cells before infusion to generate transient expression of CAR T cells in the cell product93,94. This kind of manufacturing is much simpler than conventional methods of generating cell therapies, which typically rely on viral vectors to stably incorporate the CAR transgene into the genome, leading to persistent engineering of the transferred cells and all emerging cell progeny derived from the product 58 . Ongoing clinical trials will provide evidence that can be used to assess the utility of these transiently active cell products.
While emerging preclinical and clinical data for CAR T-cell therapy in LN is promising, its full potential will become clearer as ongoing research addresses key challenges. Current and future efforts aim to enhance safety and efficacy through improved engineering techniques, developing personalized treatments based on patients’ immune profiles, and optimizing manufacturing processes for greater accessibility. Advancements in T-cell engineering and delivery methods are actively being explored and may further refine therapeutic strategies while combination therapies with biologics or small molecules could further improve treatment outcomes. In addition, advancements in biomarker discovery and immune profiling will be crucial for selecting appropriate candidates and monitoring treatment responses95,96. As research advances, T-cell therapies have the potential to revolutionize LN management, offering a more targeted and durable treatment option for patients, which is potentially curative in some.
Challenges and future directions for CAR-T therapy in LN
While CAR T-cell-based treatment for LN shows considerable promise, several logistical challenges must be addressed. Currently, all approved CAR-T therapies are manufactured using patient-derived autologous T cells as the starting material58,97. The basic workflow is summarized in Fig. 2, where cells are typically collected by leukapheresis, then expanded ex vivo, where the engineered genetic material is introduced by viral transduction58,79 or by transfection of nucleic acids encoding engineered constructs98,99. However, the use of autologous T cells inherently means that the dysfunctional state of lupus patient-derived T cells 100 could potentially affect the characteristics of the manufactured CAR-T product. Therefore, ongoing clinical trials should carefully monitor the CAR T-cell phenotype after manufacturing to compare lupus patients’ T cells to those manufactured from healthy donors or cancer patients.
A major advantage of using patient-derived material is that the cells are fully human leukocyte antigen (HLA) matched, eliminating side effects such as GvHD and rejection of the engineered cells101,102. However, the need to engineer cells for each patient has led to limitations in the adoption of CAR-T therapies due to logistical and economic factors. Specifically, patient material must be collected and sent to a specialized manufacturing facility, where the redirected CAR T-cell products are prepared. This cumbersome, specialized process has limited the availability of CAR-T products and drives up manufacturing costs 103 . To address these issues, efforts are underway to generate an “off-the-shelf” allogeneic CAR T-cell product, which would be manufactured from healthy donors and used to treat any patient, as illustrated in Fig. 2. While this approach could streamline logistics and reduce cost, none of these allogeneic products have been clinically approved104,105. Complete responses using allogeneic products have been reported for some indications although the persistence of the cells appears to be limited and long-term responses have been less impressive than for autologous products when compared in similar indications106,107. This is thought to be due largely to acute allogeneic graft rejection, which leads to rapid elimination of the therapeutic cells101,107. Given the rapid clearance of allogeneic CAR T cells, alternative approaches are being explored to improve persistence and therapeutic impact while minimizing associated risks.
T-cell-based therapies also face significant risks and toxicity concerns that must be considered in the context of LN to ensure their safety, efficacy, and accessibility in this patient population. Safety concerns emerge at several stages in CAR-T therapy. First, both autologous and allogeneic CAR T-cell approaches may require lymphodepletion prior to administration to enhance the efficacy of the infused CAR T cells108,109. Lymphodepletion is thought to create a more favorable environment for the expansion and activity of the infused T-cell product 110 . Current lymphodepleting regimens used to prepare patients for adoptive T-cell transfer utilize both cyclophosphamide and fludarabine, which carry significant risk of germ-line toxicity and are known carcinogens 111 . This is of particular concern in younger patients of reproductive age. In addition, this requirement for lymphodepletion can pose challenges in non-malignant diseases like LN, where side effects, such as increased susceptibility to infections, malignancies, and other complications, may increase the risk vs. benefit112,113.
The problems associated with the toxic lymphodepletion regimen might be addressed by engineering CAR T cells in a way that they efficiently engraft into patients without lymphodepletion. Indeed, we have observed that adoptively transferred T cells can be engrafted into lymphoreplete hosts by modifying the engineered cells with a stabilized RNA encoding a constitutively active form of Stat5 (Stat5*) 114 . Clinical studies will need to address the efficiency of Stat5* engineered CAR T cells in the context of engraftment and function, but removing chemotherapeutics would be an attractive feature of cell therapy for situations where it is contraindicated.
Next, the CAR T-cell therapy itself can cause cytokine release syndrome (CRS)62,63,115–117. CRS is characterized by excessive release of proinflammatory cytokines, including IL-6, which can lead to life-threatening systemic inflammation and is a well-documented complication associated with CAR T-cell therapies108,109,118. A related, but distinct, side effect of CAR-T therapy is immune effector cell–associated neurological syndrome (ICANS), which is less common but can be fatal in some cases119,120. Finally, off-target effects, which are difficult to predict from preclinical models, can manifest when targeting antigens that may also be present on healthy cells. Several examples of “on-target off-tumor” effects have been observed when treating cancer patients with CAR-T therapy (reviewed in Bonifant et al. 121 and Flugel et al. 122 ). These “off-target” effects are the basis of therapeutic utility of CAR-T therapy in rheumatologic disorders since CAR-T can efficiently induce B-cell aplasia. However, some of the CAR constructs proposed for use in LN including BCMA and GPRC5D have been observed to induce off-target toxicity resulting in neurological complications that are thought to result from CNS expression of these two antigens, distinct from ICANS123,124.
As experience with managing CAR-T therapies increases, clinics have improved monitoring of CRS and ICANS, which has improved outcomes by intervening early with steroids and IL-6 receptor inhibiting agents like tocilizumab 125 . Unfortunately, off-target effects are still difficult to predict and manage. Some of the approaches that have been developed to address this risk is by leveraging short-lived CAR T cells with surface markers or drug-inducible “kill switches”126–130, but the utility of these tools is yet to be determined in the clinic. Overall, while still in the early stages of development for LN, T-cell-based therapies hold tremendous promise for patients living with this difficult disease. Ongoing preclinical studies and clinical trials will help determine their role in the future of LN treatment.
Conclusion
LN presents significant challenges due to its complex pathophysiology and heterogeneous clinical manifestations. The variability in immune mechanisms and treatment responses highlights the need for personalized approaches that tailor T-cell therapies to individual patients. In addition, the manufacturing and scalability of T-cell-based treatments pose substantial hurdles, as the production process requires harvesting of patient cells, ex vivo engineering, and expansion to therapeutic doses, an effort that is labor-intensive and costly. Ensuring consistency and quality across batches remains a challenge and may limit the widespread availability of these therapies.
Despite these challenges, advancements in T-cell therapy for LN are promising, with advancements in understanding the disease mechanisms, novel therapeutic targets, and innovative technologies paving the way for new treatment options. T-cell-based therapies offer a groundbreaking opportunity to treat immune dysregulation in LN while minimizing systemic side effects, potentially transforming treatment options. Continued research, collaboration, and technical innovation are essential to overcome existing challenges and unlock the full potential of T-cell therapies, ultimately improving outcomes for patients suffering from this complex autoimmune disease.
Footnotes
Acknowledgements
Schematics were created with BioRender.com.
Ethical approval
Not applicable.
Statement of human and animal rights
This review article is based on previously published literature and does not include any original studies involving human participants or animals conducted by the authors.
Statement of informed consent
There are no human subjects in this article and informed consent is not applicable.
Authors’ contributions
MDT, RTO, and MAC each contributed to drafting and editing the manuscript.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: 2024 Lupus Research Alliance Targeted Research Program on Engineered Cell Therapies.
Declaration of conflicting interests
The author(s) declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: RTO and MDT have filed a patent application on Stat5* modified T cells.
Data availability statement
This review is based on previously published literature. All data supporting the conclusions are available in the cited sources. Schematics generated by the authors are included within the article.