
Abstract
Mucopolysaccharidosis type IIIC (MPS IIIC) is a severe neurodegenerative lysosomal storage disease caused by the loss-of-function of the lysosomal transmembrane protein acetyl-CoA: heparan-α-glucosamine N-acetyltransferase. MPS IIIC is characterized by the accumulation of the glycosaminoglycan (GAG) heparan sulfate. There is no treatment for this disease. We generated a new MPS IIIC mouse model and confirmed disease phenotypes such as GAG accumulation, splenomegaly, neurological defects, and presence of disease-specific non-reducing end carbohydrates. To explore a new therapeutic strategy for this condition, we transplanted wild-type (WT) hematopoietic stem and progenitor cells (HSPCs) into lethally irradiated 2-month-old Hgsnat−/− mice and analyzed the resulting impact 6 months later. Transplanted HSPCs differentiated into macrophages in tissues and microglia-like cells in the brain. This resulted in a partial restoration of Hgsnat expression and enzymatic activity along with a significant reduction of the MPS IIIC-specific non-reducing end carbohydrate in the treated Hgsnat−/− mice compared to untreated Hgsnat−/− mice or Hgsnat−/− mice transplanted with Hgsnat−/− HPSCs. In addition, WT HSPC transplant resulted in improved neurological defects, reduction in splenomegaly, and urine retention in the Hgsnat−/− mice. Furthermore, presence of glomerular hyaline bodies with focal fibrosis and sclerosis was observed in the kidney of the disease controls, whereas these abnormalities were improved in the Hgsnat−/− mice treated with WT HSPCs. These data support that HSPC transplantation presents a promising therapeutic avenue for MPS IIIC and represents the first step toward the clinical translation of an HSPC-mediated therapy strategy for MPS IIIC.

Keywords
Introduction
Mucopolysaccharidosis type III (MPS III), is a group of four lysosomal storage disorders (LSDs), collectively known as Sanfilippo syndrome, characterized by intra-lysosomal accumulation of the glycosaminoglycan (GAG) heparan sulfate (HS)1,2. The four sub-types (types A–D) of MPS III arise from the loss of one of the four enzymes needed to breakdown HS: sulfamidase (SGSH) is affected in MPS IIIA, α-N-acetylglucosaminidase (NAGLU) in MPS IIIB, acetyl-CoA: heparan-α-glucosamine N-acetyltransferase (HGSNAT) in MPS IIIC, and N-acetyl glucosamine-6-sulfatase (GNS) in MPS IIID 3 . A deficiency of any of these enzymes leads to an accumulation of HS in different tissues including, the brain, spleen, and liver 4 . However, the non-reducing terminal carbohydrate structure of HS is specific to each disease subtype 5 . These non-reducing terminal carbohydrates allow for disease-specific storage to be measured for diagnosis in patients and to assess therapeutic interventions in preclinical studies5–8.
MPS IIIC manifests in early childhood, normally around 3 to 6 years of age. The disease normally presents with behavioral changes, developmental milestone delay, hyperactivity, organomegaly, autistic features, and motor functions, culminating in early death due to overwhelming neurodegeneration1,3,9. MPS IIIC has a prevalence of around 0.34 per 100,000 births 10 , but recent studies have identified non-syndromic MPS IIIC patients who suffer from visual deterioration such as retinitis pigmentosa and night blindness due to loss of rod photoreceptors and severe retinal degeneration; similar ophthalmic complications are also present in the syndromic patients11–13. There is no effective therapy for this disease and current treatments only manage disease symptoms. For MPS IIIA, Adeno-Associated Virus (AAV)-mediated gene therapy has been evaluated in clinical trials; one trial involved brain injections of AACrh10 overexpressing human sulfamidase (SGSH) which has been discontinued due to safety concerns 14 , and another trial utilized intravenous administration of scAAV9-SGSH which resulted in a reduction in cerebrospinal fluid levels of HS as well as neurodevelopmental gain compared to natural history studies 15 . However, the HGSNAT enzyme affected in MPS IIIC is a lysosomal transmembrane enzyme, which adds complexity to the development of therapies such as adeno-associated virus gene therapy and enzyme replacement therapy16–19.
Bone marrow transplant has been used in patients with MPS III, and the outcomes have been mixed. For MPS IIIB, some studies reported unsuccessful prevention of neurocognitive symptoms20,21,22, and another reported possible attenuated progression of the disease in one patient 23 . In contrast, a 2.5-year-old patient affected with MPS IIIA received hematopoietic stem and progenitor cell (HSPC) transplant and showed improvement in disease phenotypes and continued cognitive development compared to six other non-treated patients with the same mutation in the N-sulfoglucosamine sulfohydrolase (SGSH) gene even 8 years later 24 . Furthermore, a recent study has shown the HSPC transplant combined with gene therapy to introduce SGSH under the control of a strong promoter may be effective for treating MPS IIIA as it led to decreased brain HS as well as significant improvement in neuroinflammation in MPS IIIA mice25,26, and is now in phase I/II clinical trial in pediatric patients with promising results so far including SGSH activity detected in cerebrospinal fluid and reduction of HS storage 27 . These studies indicate that unmodified wild-type (WT) HSPC transplantation has demonstrated limited efficacy in MPS III, whereas when combined with gene therapy, HPSC transplantation enables HSPC-derived cells to overproduce the missing enzyme, leading to improved efficacy. However, MPS IIIA and MPS IIIB are caused by mutation in soluble lysosomal enzymes in contrast to MPS IIIC that is cause by mutation in a lysosomal transmembrane protein. Indeed, while cross-correction has been extensively demonstrated in several LSDs due to defective soluble lysosomal enzymes by secretion-recapture or enzyme replacement therapy, this phenomenon remains underexplored in the context of non-secreted proteins.
We previously demonstrated that transplantation of syngeneic WT HSPC could rescue cystinosis, another LSD also caused by loss of function of a lysosomal transmembrane protein, leading to long-term preservation of the kidney, eye, and thyroid structure and function28–30. We developed an HSPC gene therapy strategy and are currently conducting a phase I/II clinical trial for an autologous, lentiviral vector (LV)–modified, CD34+ HSPC transplantation approach for cystinosis (ClinicalTrials.gov Identifier: NCT03897361). In addition, we found that a major rescue mechanism involved lysosomal cross-correction from HSPC-derived macrophages to the disease cells via tunneling nanotubes31–33. We have also previously demonstrated that WT HSPC transplantation led to long-term rescue in a mouse model of Friedreich’s ataxia, a neurodegenerative disease, and in the 5xFAD Alzheimer’s disease mice33,34. In both disease models, HSPCs engrafted and differentiated into microglia-like cells in the brain which led to the preservation of neurons as well as locomotor and neurological function33,34.
In this present study, we tested the therapeutic efficacy of WT HSPC transplant in MPS IIIC. To this end, we first generated a novel mouse model of MPS IIIC, Hgsnat−/− mice, which recapitulates key features of the disease including GAG accumulation, organomegaly, and neurological deficits. Single systemic WT HSPC transplantation in Hgsnat−/− mice led to significant phenotypical benefits, representing the first step toward a clinical translation of an HSPC gene therapy strategy for MPS IIIC.
Materials and Methods
Mice
The new mouse model of MPS IIIC was generated at Jackson Laboratory by knocking out exon 2 of the Hgsnat gene with CRISPR/Cas9 technology with the following guide RNAs Hgsnat_up_crRNA1: 5′-GCGCACATGGGGTGTTGCGC-3′, Hgsnat_down_crRNA1: 5′-AACTCGATAGAATGTGGTCA-3′, Hgsnat_up_crRNA2: 5′-CTCAGAGCACGGGCGGTCTC-3′, and Hgsnat_down_crRNA2: 5′-AAGGATAAGAGACATCCTTA-3′. The knockout was confirmed by sequencing. Mice were bred using heterozygous breeders. Mutant mice were assigned randomly to either treated, untreated, or Mock groups, WT littermates remained as controls, and heterozygous carriers were either kept on as future breeders or sacrificed depending on the colony’s needs to remain fecundity and future litters for treatment. For genotyping, tail biopsies were digested in a solution containing 200 μl of tail lyses buffer (DirectPCR, 102-T, Viagen, USA) and 3 μl of proteinase K at 55°C overnight. Samples were submitted to polymerase chain reaction (PCR) reaction with the common forward primer 5′-GTG GAC ATG GGC GAA CAA AA-3′, WT reverse primer 5′-GGG TTT CTC TGT GTA GCT CT-3′, and the mutant reverse primer 5′-TGC TGA GGA CGC GTA CAA TA-3′. Green fluorescent protein (GFP)-transgenic mice (C57B1/6-Tg (CAG-EGFP)1Osb/J; 003291, Jackson Laboratory, USA), ubiquitously expressing enhanced GFP under the control of the chicken beta-actin promoter, were used as the WT HSPC donors of HSPCs. Animals were maintained under conditions approved by the Animal Care Program (ACP) at UCSD. All procedures were approved by the Institutional Animal Care and Use Committee (IACUC). All mice were C57BL/6 background and were used at 2–10 months of age. Mice were kept in small cages provided by ACP and given continuous access to sterile food and water. Pre-established exclusion criteria were used according to IACUC guidelines and mice were assigned to treatment groups following genotyping.
HSPC Isolation, Transplantation, and Engraftment
Bone marrow cells were flushed from the femurs of 6- to 8-week-old WT, GFP-transgenic mice, one donor was usually used for two recipients. HSPCs were isolated by immunomagnetic separation using an anti-Sca1+ antibody conjugated to magnetic beads (130-123-124, Miltenyi Biotec, Germany). Sca1+ HSPCs were directly transplanted by tail vein injection of 2 × 106 cells resuspended in 100 μl of phosphate-buffered saline (14190-144, PBS) into lethally irradiated (7 Gy; X-Rad 320, PXi) MPS IIIC mice. Bone marrow cell engraftment of the transplanted cells was measured in peripheral blood at 8 months after transplantation. Heart puncture was performed following terminal anesthesia with ketamine (Ketaset, Zoetis), and blood was collected; 20 μl of whole blood was mixed by pipetting in 1-ml red blood cell lysis buffer (00-4333-57, eBioscience, USA) and following a 10-min incubation on ice. After incubation, 4-ml PBS were added and samples were centrifuged at 500 × g for 10 min. Following centrifugation, the samples were run through a BD Accuri C6 plus flow cytometer instrument to measure GFP+ cells from 10,000 live peripheral white blood cells.
Behavioral Test
Mice were tested at 10 months of age (8 months post-transplant) using two types of behavioral tests to determine gait and locomotor activity. Prior to the mice being tested, they were placed in the testing room for 30 min to acclimate to the testing environment. Open field was used to test locomotor activity using an open field arena (50 × 50 cm); the test was performed in the morning between 8 am and 12 pm. Mice were placed in the center of the arena and allowed to explore for 15 min while their movement was recorded using an automated tracking system (ANY-maze). Values generated by the ANY-maze software pertaining to measurable locomotor activity were collected and sorted based on treatment group to ascertain significant differences in the locomotor activity of each treatment and control group. The day after open field was done, CatWalk gait analysis was performed using an automated gait analysis system (CatWalk, Noldus Instruments, Netherlands); the test was performed in the morning between 8 am and 12 pm. Animals were placed at one end of the walkway (130 × 68 × 152 cm) and allowed to run down the length of the walkway, as two light sources illuminated the surface contact of paws with the glass floor, producing an image of a paw print. During locomotion, the glass walkway was filmed from below by a video camera. The CatWalk software program was used to analyze recorded footage, define individual paw prints, and give readouts of multiple parameters of gait. For each animal, four unbroken bouts of locomotion, during which animals ran down the walkway at a consistent speed, were used for analysis.
Urine Collection and Organomegaly
Urine volume residing in the bladder was collected using 1-ml Insulin Syringe (26027, Exel Comfort Point) and measured from terminally anesthetized mice with ketamine prior to cervical dislocation. Following sacrifice via cervical dislocation, tissues were collected from mice for downstream processing and analysis. The following tissues were collected for analysis: spleen, liver, kidney, eye, brain, and bladder. Spleen and liver masses were measured upon sacrifice and normalized to individual mouse body weight to detect organomegaly.
RNA Purification and Real-time Quantitative PCR
Tissues collected for genomic analysis were placed in 200 µl of RNAlater solution (AM7020, Invitrogen) and flash frozen in liquid nitrogen. Whole RNA extraction was performed using RNeasy Mini Kit as per manufacturer protocol (74106, Qiagen, Germany). cDNA synthesis occurred by reverse transcription (RT) of 10–15 mg tissue RNA using iScript cDNA Synthesis Kit (1708890, Bio-Rad, USA). RT-qPCR reaction assembly consisted of 10 μl iTaq Universal SYBR Green (1725121, Bio-Rad), 5 μl of 1:10 diluted cDNA (5 ng/µl), 0.4 μl forward and reverse primer (10 μM), and 4.6 μl H2O, and ran on a LightCycler 480 ii (Roche, Basel, Switzerland) under the following conditions: 95°C (30 s); 40 cycles of 95°C (5 s) and 60°C (30 s); then 65°C (5 s); and 95°C (5 s). Gene expression was measured using the delta/delta CT method relative to MPS IIIC and normalized to glyceraldehyde 3-phosphate dehydrogenase (Gapdh).
HGSNAT Enzyme Activity
Enzymatic activity of the HGSNAT enzyme was assayed from flash frozen tissues extracted from mice that were not perfused as previously described
35
. Tissues were homogenized in 100 µl of MilliQ water containing protease inhibitor cocktail (P8340, Millipore Sigma, USA) using the Precellys 23 Touch Homogenizer. The total protein content of the homogenized tissue was quantified using Pierce BCA protein (23205, Thermo Fisher Scientific, USA) kit following manufacturer protocol. The samples were then made up to 10 µl in MilliQ water containing 30 µg of protein. The samples were then combined with 10 µl of McIlvain Buffer (pH 5.7) containing 3 mM of 4-methylumbelliferyl-β-
Histopathology
Dissected tissues intended for histology staining were fixed in 10% buffered formalin (77507-018, VWR, USA) for 24 h and placed in 70% ethanol prior to being transferred to UCSD Moores Cancer Center Biorepository & Tissue Technology Center. Tissues were embedded in paraffin, sectioned at 4 μm, and stained with hematoxylin & eosin (H&E) staining to assess histological abnormalities. Kidney tissues were further analyzed to assess the degree of cortical damage and the number glomerular cells was quantified. Dr Valeria Estrada (Tissue Technology Shared Resource, Moores Cancer Center, UCSD) supervised the processing of tissues following fixation up to staining and provided a blinded histopathology report. Our team then quantified the number of normal and damaged glomeruli blindly in the whole kidney section stitched images based on the size and morphology of the glomeruli. Alcian blue staining was also performed at pH 1 in order to stain sulfated mucins such as HS.
Neurofilament Light Chain ELISA
Mice serum was collected by retro-orbital eye bleed prior to sacrifice using Micro-Hematocrit Capillary Tubes (22-362-566, Fisher Scientific, USA). Briefly, approximately 1 ml of mouse blood was collected, incubated at room temperature for 20 min, then centrifuged at 6,000 × g for 15 min at 4°C, and the supernatant was collected. The serum was then stored at −80°C. Neurofilament light chain (NfL) presence in the serum was measured using the NF-L ELISA Kit (NB013370, Novus Biologicals, USA) following manufacturer’s protocol and read on a VersaMax Turntable Microplate Reader (Molecular Devices, USA).
Immunofluorescence Staining, Image Acquisition, and Analysis
Tissues were fixed in 4% paraformaldehyde (J19943-K2, Thermo Fisher Scientific) for 24 h, then transferred to 10% sucrose, 20% sucrose, and 30% sucrose (S0389, Millipore Sigma) for 24 h respectively in 4°C before being embedded in Tissue-Tek Optimal Cutting Temperature buffer at −80°C (4583, Sakura Finetek, USA). Tissues then were sectioned in cryostat at a thickness of 12 µm, unmasked using citrate unmaking solution (147465, Cell Signaling Technology) through pre-warm antigen retrieval solution at 95°C for 10 min, cooled at room temperature for 15 min, and incubated in blocking solution comprised of 0.25% Triton X-100 (X100, Millipore Sigma) and 3% BSA (A3294, Millipore Sigma) in tris-buffered saline (351-086-131, Quality Biological, USA). Sections were then incubated overnight at 4°C with the following primary antibodies: chicken anti-GFP (1:200; AB290, Abcam), anti-CD68 (1:50, AB5344, Abcam), anti-IBA1 (1:500, Synaptic Systems), anti-GFAP (1:250, 50-197-5703, Fisher Scientific), and CD14 (1:250, ab221678, Abcam, United Kingdom). The appropriate Alexa Fluor–conjugated secondary antibodies (1:1,000, A21247, PIA32728, PIA32733, A11039, Fisher Scientific) were used for the visualization of antigens. Images were captured by a Keyence microscope and Nikon Eclipse Ti2 microscope and analyzed with the ImagePro software (BitPlane, Oxford Instruments, UK). The fluorescence area was quantified from 9 × 9 40× stitched images per sample using Image-Pro 10 software (Media Cybernetics, USA).
Glycan Reductive Isotope Labeling Liquid Chromatography/Mass Spectrometry
Glycan Reductive Isotope Labeling Liquid Chromatography/Mass Spectrometry (Grill LC/MS) was carried out as previously described in Lawrence et al’s study5,36. Briefly, mouse tissues were flash frozen in liquid nitrogen and lyophilized to obtain the dry weight, based on which the concentration of the pronase in the digestion buffer was calculated. The samples were then incubated with the pronase (P5147, Millipore Sigma) on the shaker in 37°C for 24 h and subsequently in 60°C for 48 h. GAGs were eluted by loading the supernatant into Poly-Prep Chromatography columns (731-1550, Bio-Rad) filled with DEAD Sephacel beads (16550, Millipore Sigma) and then into the PD-10 desalting columns (17085101, Cytiva, USA). After the elution, GAGs were lyophilized into powder state and treated with DNase (EN0521, Thermo Fisher Scientific) and Chondroitinase ABC (C2905, Millipore Sigma) for further purification, followed by beta-elimination via incubation in NaOH overnight. The resulting solution was eluted through the two types of chromatography mentioned above again to enhance the purification, and the isolated GAGs were treated with a mixture of heparan lyases I, II, and III (50-008, 50-011, 50-012, Ibex, USA) to liberate biomarker units. After being digested with the heparan lyases, GAGs samples were spiked with standards tagged with [13C6]Aniline (Millipore Sigma). These were run through LC/MS using a LTQ Orbitrap Discovery electrospray ionization mass spectrometer (Thermo Scientific) equipped with quaternary high-performance liquid chromatography pump (Finnigan Surveyor MS pump) and a reverse-phase capillary column. The resulting peaks were quantified based on the known amount of the standards.
Statistical Analysis
We did not exclude any animals from our experiments. Experimenters were blinded to the genotype and treatment group of the specific sample to every extent possible. In all experiments, cohorts were age-matched. Non-objective computational measurements such as immunostaining quantification by scanning devices did not require blinding. All data are presented as the mean ± standard error of the mean (SEM), and the statistics and graphs were performed using GraphPad software Prism v.9.0. One-way analysis of variance followed by Tukey’s multiple comparisons was used to compare multiple groups unless the data were not normally distributed, where a Kruskal–Wallis followed by Dunn’s multiple comparison. Statistical significance was set at *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001.
Results
Novel Mouse Model of MPS IIIC Accumulates Disease-Specific Non-reducing End Carbohydrates
To investigate the potential of WT HSPC transplantation for treating MPS IIIC, we first generated a novel MPS IIIC mouse model by removing exon 2 of the murine Hgsnat gene in a C57/BL6 genetic background, leading to a frameshift and early stop codons in exons 3 and 4 (Fig. 1A). This mouse model was generated due to the lack of commercially and readily available murine model of MPS IIIC. Hgsnat gene knockout was confirmed by sequencing (data not shown) and PCR (Fig. 1B). Curiously, breeding of heterozygote Hgsnat mice did not follow a Mendelian ratio, with less heterozygous mice than expected and more WT mice (chi-square = 17.974 with 2 degrees of freedom; P = 0.0001, n = 608). The quantity of knockout Hgsnat−/−mice was as expected, and no difference in life span was observed at least up to 15 months of age (latest time point studied). We showed that the Hgsnat−/−mice accumulate the MPS IIIC disease-specific non-reducing end carbohydrates that appear in the LC/MS at 752, 832, and 961 (Fig. 1C) with the possible structure outlined in Fig. 1D and previously reported by Lawrence and colleagues 5 . These results validate our model as MPS IIIC, and its full characterization is depicted in Fig. 1.
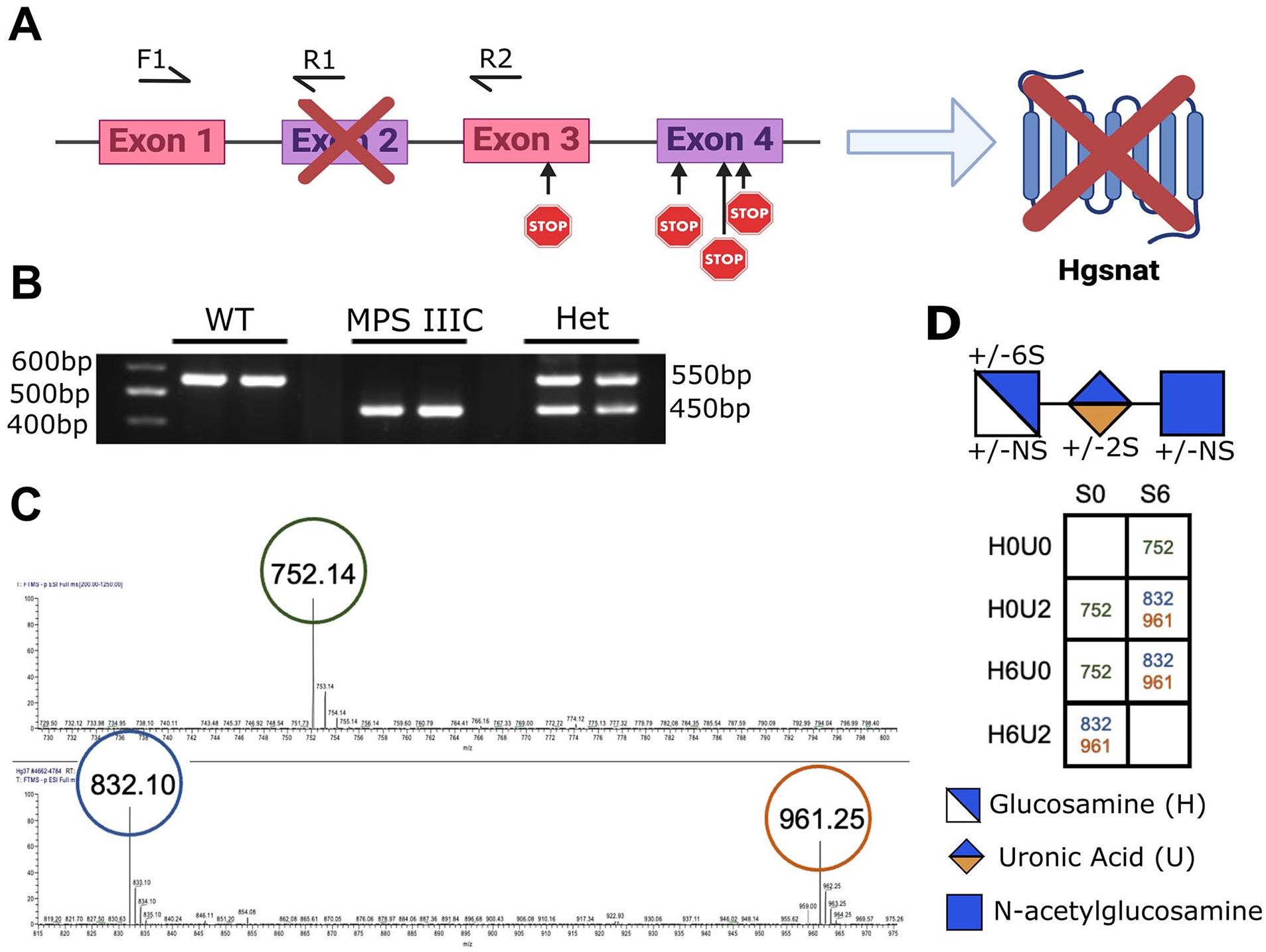
Generation of a novel MPS IIIC mouse model: (A) Schematic representation of the generation of a novel MPS IIIC mouse model. CRISPR/Cas9-mediated knockout of exon 2 in Hgsnat caused a frameshift introducing early stop codons in exons 3 and 4 leading to the absence of protein. (B) Genotyping of WT, MPS IIIC (Hgsnat−/−), and heterozygous (Hgsnat+/-) mice was performed by PCR analysis from genomic DNA. The location of the primers is depicted in (A). F1 is a common forward primer for both WT and mutant alleles, R1 is the WT primer, and R2 is the mutant reverse primer. (C) Liquid chromatography/mass spectrometry chromatographs of 12 C6 aniline-tagged non-reducing end MPS IIIC disease-specific trisaccharide. (D) Displayed geometric symbols of the NRE carbohydrate structure and table showing the theoretically possible trisaccharide structure for MPS IIIC.
Transplantation of WT HSPCs Restores Partial Hgsnat Expression and Activity in Tissues and Decreases the Disease-Specific Non-reducing End Carbohydrate in the Brain
To assess the impact of WT HSPC transplantation on MPS IIIC, we intravenously injected lethally irradiated 6- to 8-week-old Hgsnat−/−mice with Sca1+ HSPCs isolated from WT GFP-transgenic mice (Test, n = 12; 8 females, 4 males). As controls, we analyzed WT littermates (WT, n = 14; 8 females, 6 males), non-treated Hgsnat−/− mice (MPS IIIC, n = 11; 7 females, 4 males), and lethally irradiated Hgsnat−/−mice transplanted with Sca1+ Hgsnat−/− HSPCs (Mock, n = 6; 2 females, 4 males). These mice were transplanted at 2 months of age and sacrificed 8 months later at 10 months of age for analysis (Fig. 2A). Donor-derived GFP+ HSPC engraftment was measured in the peripheral blood of the Test mice by flow cytometry at 8 months post-transplant and ranged from 11% to 89.7% with an average of 53% engraftment (Table 1); variations in engraftment may come from tail vein injection efficiency, and differences in weight, which can affect irradiation exposure. We assessed engraftment of the WT HSPC–derived cells in tissues by measuring Hgsnat expression using quantitative PCR. Partial restoration of Hgsnat expression was observed in all the tested tissues with a recovery of HGSNAT expression in the Test mice compared to WT of 0.5% in the brain, 4% in the kidney, and 33% in the spleen (Fig. 2B). Similarly, presence of HGSNAT enzyme activity, as assessed by a fluorometric 4-methylubelliferone assay, was observed in tissues following WT HSPC transplantation in Hgsnat−/− mice as opposed to non-treated and Mock Hgsnat−/− mice (Fig. 2C). The level of enzyme recovery in the Test mice compared to WT was 17% in the brain, 3% in the kidney, and 27% in the spleen in which HSPC engraftment is the highest (Fig. 2C).

Transplantation of WT HSPCs leads to Hgsnat expression and activity in tissues. (A) Schematic representation of the experimental design and timeline. (B) Hgsnat mRNA expression measured by qPCR in the brain, spleen, and kidney. (C) HGSNAT enzyme activity measured using a 4-methylumbelliferone fluorescent plate assay. (D, E) LC/MS analysis of 12 C6 aniline-tagged saccharides was used to quantify total heparan sulfate (HS) in the brain and kidney. The relative levels of the MPS IIIC disease-specific non-reducing end 832 trisaccharide. For the Test mice, white dots indicate mice with peripheral blood engraftment below 50% and black dots above 50%. Data are means ± SEM. P-values are denoted as follows: *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001.
Outline of Test Mice Group Gender and Percentage HPSC Engraftment.

To evaluate the impact of WT HPSC on the pathological storage, we quantified total HS and the presence of the disease-specific non-reducing end carbohydrate in the brain and kidney by Grill LC/MS. An increase in total HS was noted in the MPS IIIC and Mock mice compared to WT controls in both tissues (Fig. 2D). A significant decrease was observed in the brains of the Test mice compared to the Mock mice, while a trend toward reduction was noted in the kidneys (Fig. 2E). Noticeably, the MPS IIIC–specific non-reducing end carbohydrate 832 was increased in both untreated and Mock groups compared to WT, and significantly decreased in the Test group compared to both non-treated Hgsnat−/− and Mock mice in both tissues (Fig. 2D, E).
Transplantation of WT HSPCs Improves Locomotor Function in the MPS IIIC Mouse Model
To assess the neurological dysfunction in the Hgsnat−/− mice, we performed behavioral testing in 10-month-old mice. We tested the locomotor activity of the mice in an open field. General motor function as expressed as mean speed and distance was significantly increased in the Test mice compared to the untreated MPS IIIC and Mock mice for mean speed, and compared to MPS IIIC group for distance (Fig. 3A). Other parameters such as time spent in the center, thigmotaxis, and freezing time did not show any significant difference between the groups (data not shown). Alterations in gait and locomotor function were demonstrated using the CatWalk testing apparatus in both the MPS IIIC and Mock groups, which displayed an inability to properly couple the movement of the right front paw with the left hind paw as well as defects in support diagonal, run average speed, duration, and run duration as compared to WT littermates (Fig. 3B and Supplemental Fig. 1). In contrast, performance of the Test mice was similar to WT mice with significantly better or non-significant trends of improvement compared to MPS IIIC and Mock mice in most of the tests (Fig. 3B). Additionally, assessment of individual paws by CatWalk gait analysis demonstrated a decrease in stride length and alterations in stance, and body speed in the MPS IIIC and Mock groups compared to the WT mice. In contrast, the Test mice behaved like WT mice with significantly better or non-significant trends of improvement compared to MPS IIIC and Mock (Fig. 3B and Supplemental Fig. 1). Altogether, these data demonstrate that MPS IIIC mice exhibit locomotor impairments caused by neurological defects, as well as potential physical abnormalities such as organomegaly and urine retention as described below. These impairments are alleviated following transplantation of WT HPSCs.
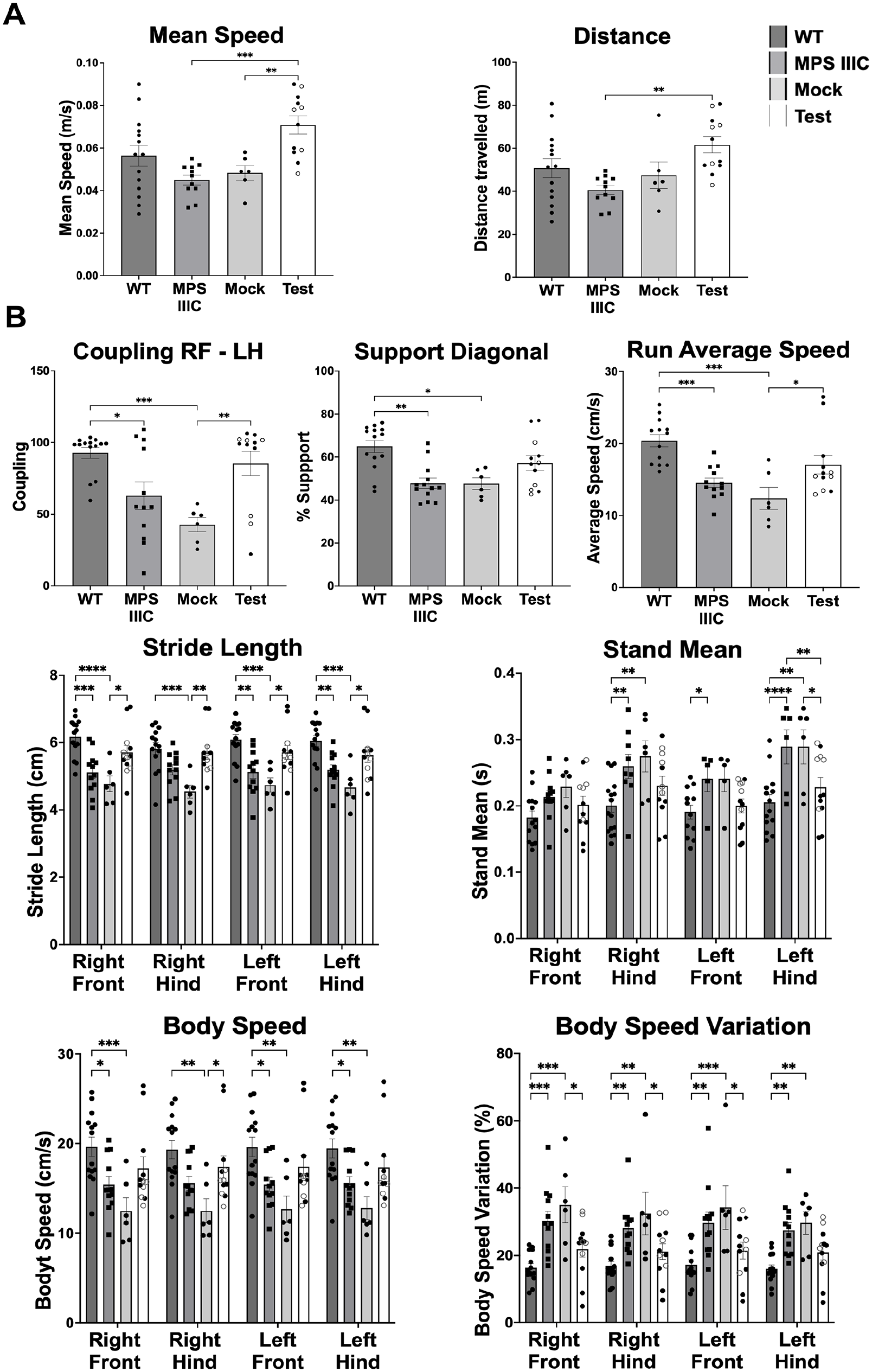
Transplantation of WT HSPC improves locomotor dysfunction in the Hgsnat−/−mice. (A) Open field test performed on 10-month-old mice MPS IIIC mice. (B) CatWalk gait analysis was used to assess locomotor function. For the Test mice, white dots indicate mice with peripheral blood engraftment below 50% and black dots above 50%. Data are means ± SEM. P-values are denoted as follows: *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001, ****P ≤ 0.001.
Transplantation of WT HSPC Ameliorates Urine Retention, Organomegaly, and Kidney Pathology in the MPS IIIC Mouse Model
Previous studies on MPS IIIA, B, and C have reported urine retention and distended bladder in the mouse models4,37,38. We confirmed the presence of urine retention and pathologically enlarged bladder in our MPS IIIC mice as well as in the Mock mice (Fig. 4A). In contrast, bladder size was decreased, and urine volume significantly reduced in the Test mice compared to MPS IIIC mice (Fig. 4B). Splenomegaly is a common feature reported in MPS III mice and patients1,39. Significant increase in spleen mass was observed in MPS IIIC and Mock mice compared to WT littermates while significant decrease was observed in Test mice compared to MPS IIIC and Mock mice (Fig. 4C). Hepatomegaly was also observed in the MPS III and Mock groups but was not improved in the Test group (Fig. 4D).

Transplantation of WT HSPC decreased urine retention, splenomegaly, and kidney pathology. (A) Images of mice bladders at sacrifice. (B) Urine volume as measured at sacrifice. (C) The size of the spleen was measured by weight as a percentage of body mass. (D) The size of the liver was measured by weight as a percentage of body mass. (E) Histopathology analysis of kidney sections stained by H&E. (F) Quantification of the damaged glomeruli in the different group of mice. (G) Alcian blue staining of kidney performed at pH 1 to stain-sulfated mucins include heparan sulfate. For the Test mice, white dots indicate mice with peripheral blood engraftment below 50% and black dots above 50%. Data are means ± SEM. P-values are denoted as follows: *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001, ****P ≤ 0.0001.
Brain, bladder, kidney, and spleen tissues were analyzed blindly for histopathological changes by a pathologist using H&E staining, and only the kidney exhibited marked abnormalities in the Hgsnat−/− mice. Specifically, in both untreated and Mock-treated Hgsnat−/− mice, the kidney exhibited glomerular hyaline bodies with focal fibrosis and sclerosis as well as some dilated tubules and vascular spaces (Fig. 4E). The number of damaged glomeruli was quantified and were significantly decreased compared to the MPS IIIC mice and showed non-significant trend compared to Mock mice (Fig. 4F).
To characterize the hyaline bodies in the kidney of the disease mice, we performed Alcian blue staining which is a polyvalent basic dye known to stain sulfated mucins such as HS 40 . Interestingly, Alcian blue stained primarily the glomeruli even in the WT mice (Fig. 4G), which is not surprising as HS proteoglycans are known to be a major component of the glomeruli and have an important role in glomerular organization and function 41 . Diffuse blue staining was also observed in the MPS IIIC and Mock groups, but also the presence of vacuoles, some of them containing GAGs (Fig. 4G). In contrast, the blue staining in kidney sections from Test mice was reduced, making them appear more similar to WT mice, with no visible vacuoles (Fig. 4G).
Transplantation of WT HSPC Differentiated Into Microglia-Like Cells in the Brain and Macrophages in Peripheral Tissues
We characterized the engraftment and differentiation of the GFP+ HSPCs in the brain, which were immunoreactive with Iba1 identifying these cells as microglia-like cells (Fig. 5A). We also explored tissue inflammation by examining CD68 expression which is associated with macrophages/microglia activation and phagocytic activity42,43. CD68 expression was significantly increased in the brain of untreated MPS IIIC and Mock mice compared to WT mice (Fig. 5B). In contrast, reduction of CD68 expression was observed in the Test mice compared to MPS IIIC and Mock mice while significance was reached compared to MPS IIIC only (Fig. 5B). In addition, neuronal damage was investigated by measuring NfL levels in the serum44,45. A significant increase in NfL levels was observed in the MPS IIIC and Mock mice compared to the WT mice, which was rescued in the Test mice (Fig. 5C). Astrogliosis was also tested using anti-GFAP staining showing a non-significant increase in MPS IIIC and Mock mice, and decrease in Test mice (Supplemental Fig. 2).

WT HSPC–derived cells engraft and differentiated into microglia-like cells in the brain and macrophages in tissues. (A) Representative immunofluorescence image of brain sections from WT, MPS IIIC, Mock, and Test mice stained with anti-GFP (green) and anti-Iba1 (red) antibodies. Scale bars, 40 μm. (B) Representative immunofluorescence image and area analysis quantification of brain sections from WT, MPS IIIC, Mock, and Test mice stained with anti-GFP (green) and anti-CD68 (red) antibodies. Scale bars, 100 µm. (C) NfL levels in the serum were measured by ELISA. (D) Representative immunofluorescence image and area analysis quantification of kidney sections from WT, MPS IIIC, Mock, and Test mice stained with anti-CD68 (red) antibodies. Scale bars, 100 µm. Quantification of CD68-positive staining. For the Test mice, white dots indicate mice with peripheral blood engraftment below 50% and black dots above 50%. Scale bars, 300 μm. Data are means ± SEM. P-values are denoted as follows: ***P ≤ 0.001, ****P ≤ 0.0001.
We also characterized the engraftment and differentiation of GFP+ HSPCs in peripheral tissues including liver and kidney and found substantial engraftment of GFP+ HSPC–derived cells in these tissues (Fig. 5D and Supplemental Fig. 2). These cells were immunoreactive for CD14, identifying them as macrophages (Fig. 5D and Supplemental Fig. 2). Significant increase of CD14+ cells was seen only in Mock mice compared to WT probably because of the toxicity of irradiation without the positive impact of the healthy cells. In addition, significant increase of CD68-positive cells was measured in the kidney and liver from the MPS IIIC and Mock mice compared to both WT and Test mice (Fig. 5D and Supplemental Fig. 2), suggesting an increase of inflammation in the kidney and liver in the disease mice and improvement following WT HSPC transplant.
Discussion
In the present study, we explored the therapeutic potential of WT HSPC transplant for the multisystemic lysosomal storage disease MPS IIIC. MPS IIIC is caused by mutations in the HGSNAT gene encoding a lysosomal transmembrane enzyme. Though other MPS diseases, due to mutations in gene that encode secreted enzymes, have the potential to be treated with AAV delivery17–19,46 or enzyme replacement therapy 47 , these approaches are less effective for diseases caused by mutations in genes that encode transmembrane proteins, such as MPS IIIC 46 . Furthermore, pre-clinical studies have had limited success using AAV delivery of HGSNAT in mice models of MPS IIIC 46 and have only targeted the brain by intraparenchymal convection of AAV2-HGSNAT 48 . Another study has explored a novel rAAVhHGSNATEV that contains an extracellular vesicle (EV)-mRNA-packing signal to promote HGSNAT expression and packaging into EVs, but this has only been tested in vitro so far 49 .
Unlike AAV vectors, WT HPSCs have the ability to engraft at high level in all tissues after a single injection, as they are not restricted by dose limitations or tissue accessibility as seen in human clinical trials 50 . HSPCs then differentiate into macrophages in peripheral organs and microglia-like cells in the central nervous system (CNS) as we previously demonstrated in cystinosis, Friedreich’s ataxia, and Alzheimer’s disease models29,33,34,51. In addition, the HSPC-derived phagocytic cells can deliver healthy lysosomes carrying the transmembrane lysosomal protein to the disease cells as we demonstrated in cystinosis 31 . This process resulted in long-term tissue rescue in cystinosis28–30, making it an appealing strategy for MPS IIIC. We confirmed in the present study the transplanted WT HSPC migration and differentiation into microglia-like cells in the brain and into macrophages in other peripheral tissues such as kidney in the Hgsnat−/− mice. Another study has also shown that WT HSPC is a promising therapeutic avenue for MPS IIIC 52 . The mouse model used in this study exhibited several phenotypes similar to our model, including urine retention, hepatomegaly, splenomegaly, and increased CD68 expression in tissues. However, their model displayed hyperactivity, which we did not observe. This discrepancy may be explained by differences in the age at which behavioral assessments were performed (6 vs 10 months of age). Notably, they did not observe corrections in splenomegaly or brain HS storage, unlike our study. This discrepancy may be attributed to differences in the level of HSPC engraftment, which they did not quantify. Another possible explanation is the use of a knock-in mouse model in the Pan et al’s 52 study, which expresses misfolded HGSNAT that may itself be pathogenic and limit the effectiveness of HSPC transplantation. Nonetheless, both studies support the potential of HSPC transplantation as a promising therapeutic strategy for MPS IIIC.
Representative animal models are crucial in the understanding of human diseases and for the development of novel therapies. In this study, we described a novel mouse model of MPS IIIC, which showed no activity of the HGSNAT enzyme in tissues and presented with key phenotype features of MPS IIIC disease. The hallmark of all the Sanfilippo diseases including type C is the storage of the GAG HS1,5,39. Using the Grill LC/MS method, we confirmed the accumulation of HS within the brain of the MPS IIIC mice as well as the presence of disease-specific non-reducing end carbohydrates 5 in the liver and brain. In addition, this new MPS IIIC model presents similar phenotype to others previously made such as splenomegaly, distended bladder, behavioral anomalies, and neuroinflammation4,53. Transplantation of WT HSPCs led to partial restoration of the HGSNAT enzymatic activity in the tissues and the reduction of HS and disease-specific non-reducing end carbohydrates in the brain of the Hgsnat−/− mice, showing the positive impact of the Hgsnat-expressing HSPCs in MPS IIIC. It is important to note that HGSNAT enzyme activity was measured in tissues collected from mice that were not perfused, so leukocytes and other blood-derived cells could be present, potentially interfering with the enzyme activity measurement.
Patients affected with MPS IIIC develop progressive and severe neurodegenerative disease that eventually leads to loss of speech and motor function 9 . Mouse models of MPS IIIA and IIIC have shown altered locomotor activity53,54. We have confirmed that our mouse model of MPS IIIC also displays locomotor and neurocognitive defects such as abnormal paw coupling while walking, abnormal support diagonal, decreased stride length, and increase in body speed variation. Similar locomotor defects have been reported in another MPS IIIC mouse model 53 . Importantly, we demonstrated that a single WT HSPC transplantation led to an amelioration of these locomotor deficits. This improvement may result from the reduction of the pathological HS storage in the brain, but also from the differentiation of the WT HSPC into microglia-like cells that leads to the reduction of neuroinflammation as shown in this present study but also in MPS IIIA, IIIC, and Alzheimer’s disease26,34,52. The improvement in locomotor defects in the Hgsnat−/− mice following HSPC transplant could also be due to the decrease in microglia activation and inflammation in the CNS. Indeed, macrophage/microglial activation and neuroinflammation have been shown to contribute to neurodegeneration55–57 and have been reported to present MPS IIIA 58 , IIIB58–60, and IIIC4,53,61. Here we showed increased expression of CD68 in the brain of the untreated and Mock-treated Hgsnat−/− mice, which has shown to increase in microglial cells when stimulated by inflammatory cytokines 62 . In addition, significant increase of serum NfL, a marker for neuronal damage44,45, was observed in the untreated and Mock-treated Hgsnat−/− mice compared to WT mice. We demonstrated that both CD68 expression and NfL decreased in the Hgsnat−/− mice treated with WT HSPC. Therefore, WT HSPC transplant therapeutic benefits in the CNS may come from the restoration of the HGSNAT in the neurons but also because of the decrease of neuroinflammation as previously shown in mouse models of MPS IIIA and IIIB26,63.
An interesting anomaly found in other Sanfilippo mice is urine retention and distended bladder reported in the mouse models of MPS IIIA, IIIB, and IIIC4,37,38. We also observed an increase in urine volume and distended bladder in our Hgsnat−/− mice as reported in another MPS IIIC mouse model 4 . The cause of urine retention is not clear and the lesion may result from infiltration of inflammatory cells throughout the entire urinary tract that could potentially lead to obstructive uropathy or disrupt the neurogenic aspect of micturition 37 . Interestingly, we showed that WT HSPC transplantation ameliorated this defect in Hgsnat−/− mice, possibly due to the reduction of inflammatory cells into the urinary tract as observed in the kidney. Urine retention could be also due to glomerular disease 37 . Indeed, our study demonstrates the presence of kidney pathology in MPS IIIC such as glomerular hyaline bodies with focal fibrosis and sclerosis as well as some dilated tubules and vascular spaces. Similarly, a previous study showed histological alteration in the kidney in MPS IIIC mouse model 64 . HS is known to be important for glomeruli function as it is key in maintaining the selective permeability of the glomerular basement membrane 41 . Therefore, accumulation of HS may lead to morphological and functional changes in the glomeruli of the kidney. We also showed increased inflammation in the untreated and Mock-treated Hgsnat−/− mice compared to WT mice. The histopathology defects observed in the Hgsnat−/− kidney appeared to be improved following WT HSPC transplantation, showing direct impact of the HSPC-derived macrophages in kidney rescue. Similarly, we previously showed that WT HPSC transplantation improved kidney function, improved structure, and decreased inflammation in the kidney in a cystinosis mouse model 65 .
Our data on the impact of WT HSPCs in MPS IIIC are promising as they lead to the improvement of several complications including neurological and kidney defects as well as to the reduction of the disease-specific non-reducing end carbohydrates in the brain. However, the recovery of the expression of Hgsnat and its enzyme activity was limited in tissues. HPSC transplant has been explored for other MPS diseases and while MPS I can be rescued by WT HSPC transplantation, bone marrow transplantation was not always able to significantly improve or stabilize cognitive function in MPS IIIA patients 21 . Similarly, bone marrow transplant in MPS IIIA murine neonates did not reduce HS storage in the brain, but the donor cell engraftment was low (~20%) 66 . The hypothesis for these results was that SGSH was not sufficiently expressed in HSPC-derived macrophages/microglia in tissues. Further studies support this hypothesis as MPS IIIA adult mice showed that both WT HSPC and LV-transduced MPS IIIA HSPCs, with SGSH expression driven under a spleen focus-forming virus promoter, were effective at reducing GM2 gangliosides and neuroinflammation 26 , but not in reducing HS or improving behavior 26 . In contrast, LV-SGSH-transduced HSPCs that led to ubiquitous over-expression (phosphoglycerate kinase promoter) resulted in normalization of HS, and significant reduction in neuroinflammation and improvement of behavior26,67. Similarly, the same group showed that MPS IIIA HSPCs transduced with a LV-SGSH, carrying the myeloid-specific promoter CD11b, were successful in rescuing brain HS, GM2 gangliosides, and neuroinflammation 67 . A clinical trial is now ongoing for MPS IIIA using this approach with promising clinical outcome (ClinicalTrials.gov Identifier: NCT04201405). We believe that similar to MPS IIIA, the HGSNAT enzyme is not well expressed in HSPC-derived cells leading to a significant impact of the WT HSPCs, and HSPC gene therapy should lead to even greater therapeutic beneficial the outcomes.
Our findings demonstrate WT HSPC transplantation led to the improvement of disease phenotypes including locomotor defects, urine retention, kidney pathology, splenomegaly, as well as a reduction in disease-specific non-reducing end carbohydrate despite recovery of HGSNAT enzyme activity in tissues. Therefore, these results are promising and support the development of an autologous transplantation of gene-modified HSPCs to drive a strong and ubiquitous expression of HGSNAT.
Supplemental Material
sj-docx-1-cll-10.1177_09636897251323966 – Supplemental material for Transplantation of Wild-Type Hematopoietic Stem and Progenitor Cells Improves Disease Phenotypes in a Mucopolysaccharidosis IIIC Mouse Model
Supplemental material, sj-docx-1-cll-10.1177_09636897251323966 for Transplantation of Wild-Type Hematopoietic Stem and Progenitor Cells Improves Disease Phenotypes in a Mucopolysaccharidosis IIIC Mouse Model by Rafael A. Badell-Grau, Kasra Pakravesh, Kevin Eric Thai, Frankie Son, Rola Chen, Joseph Rainaldi, Kalvin Duong, Pauline Losay, Anusha Sivakumar, Veenita Khare, Alexis N. Corl, Rushil Pithia, Christine Tran, Jefferey D. Esko and Stephanie Cherqui in Cell Transplantation
Supplemental Material
sj-tiff-2-cll-10.1177_09636897251323966 – Supplemental material for Transplantation of Wild-Type Hematopoietic Stem and Progenitor Cells Improves Disease Phenotypes in a Mucopolysaccharidosis IIIC Mouse Model
Supplemental material, sj-tiff-2-cll-10.1177_09636897251323966 for Transplantation of Wild-Type Hematopoietic Stem and Progenitor Cells Improves Disease Phenotypes in a Mucopolysaccharidosis IIIC Mouse Model by Rafael A. Badell-Grau, Kasra Pakravesh, Kevin Eric Thai, Frankie Son, Rola Chen, Joseph Rainaldi, Kalvin Duong, Pauline Losay, Anusha Sivakumar, Veenita Khare, Alexis N. Corl, Rushil Pithia, Christine Tran, Jefferey D. Esko and Stephanie Cherqui in Cell Transplantation
Supplemental Material
sj-tiff-3-cll-10.1177_09636897251323966 – Supplemental material for Transplantation of Wild-Type Hematopoietic Stem and Progenitor Cells Improves Disease Phenotypes in a Mucopolysaccharidosis IIIC Mouse Model
Supplemental material, sj-tiff-3-cll-10.1177_09636897251323966 for Transplantation of Wild-Type Hematopoietic Stem and Progenitor Cells Improves Disease Phenotypes in a Mucopolysaccharidosis IIIC Mouse Model by Rafael A. Badell-Grau, Kasra Pakravesh, Kevin Eric Thai, Frankie Son, Rola Chen, Joseph Rainaldi, Kalvin Duong, Pauline Losay, Anusha Sivakumar, Veenita Khare, Alexis N. Corl, Rushil Pithia, Christine Tran, Jefferey D. Esko and Stephanie Cherqui in Cell Transplantation
Supplemental Material
sj-tiff-4-cll-10.1177_09636897251323966 – Supplemental material for Transplantation of Wild-Type Hematopoietic Stem and Progenitor Cells Improves Disease Phenotypes in a Mucopolysaccharidosis IIIC Mouse Model
Supplemental material, sj-tiff-4-cll-10.1177_09636897251323966 for Transplantation of Wild-Type Hematopoietic Stem and Progenitor Cells Improves Disease Phenotypes in a Mucopolysaccharidosis IIIC Mouse Model by Rafael A. Badell-Grau, Kasra Pakravesh, Kevin Eric Thai, Frankie Son, Rola Chen, Joseph Rainaldi, Kalvin Duong, Pauline Losay, Anusha Sivakumar, Veenita Khare, Alexis N. Corl, Rushil Pithia, Christine Tran, Jefferey D. Esko and Stephanie Cherqui in Cell Transplantation
Footnotes
Acknowledgements
The authors gratefully acknowledge Dr Thomas Hnasko and his team, for sharing behavioral room. They acknowledge Nathan Benmergui and Thibaut Losay for their contribution to the successful competition of this work. They also like to acknowledge Dr Biswa P. Choudhury and UCSD GlycoAnalyticsCore.
Statements and Declarations
Supplemental Material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.