
Abstract
Rheumatoid arthritis (RA) is a systemic, chronic inflammatory disease characterized by altered levels of inflammatory cytokines. One of the key cytokines involved in the pathogenesis of RA is tumor necrosis factor α (TNF-α), which plays a crucial role in the differentiation of T cells and B cells and serves as a primary trigger of inflammation and joint damage in RA. Human umbilical cord-derived mesenchymal stem cells (hUC-MSCs) have shown potential in alleviating the symptoms of RA. Previous in vitro studies indicate that TNF-α secreted by T cells can activate NF-κB in human MSCs, thereby triggering the immunoregulatory capacity of MSCs in a manner dependent on tumor necrosis factor receptor 1 (TNFR1). Inspired by these findings, we aimed to evaluate whether TNFR1 determine the therapeutic effects of hUC-MSCs on RA. First, we investigated whether TNFR1 is necessary for hUC-MSCs to inhibit TNF-α production of PBMCs, a source of elevated TNF-α in patients. Through coculture experiment, we confirmed that this inhibition was dependent on TNFR1. Subsequently, we administered hUC-MSCs or siTNFR1-MSCs to DBA/1J male mice with collagen-induced arthritis. The results indicated that hUC-MSCs significantly alleviated the pathological features of RA and suppressed the inflammatory cytokines IFN-γ, TNF-α, and IL-6 in peripheral blood, also in a manner dependent on TNFR1 either. Given the dramatic pathologic differences between hUC-MSCs and siTNFR1-MSCs treatments, we questioned whether production of growth factors and chemokines was significantly influenced by TNFR1. Consequently, we stimulated hUC-MSCs or siTNFR1-MSCs through IFN-γ, TNF-α, and IL-6, and profiled growth factors and chemokines in serum, which revealed significant changes of hepatocyte growth factor (HGF) and keratinocyte growth factor (KGF), as well as chemokines CXCL9, CXCL10, IL-8, and RANTES. In summary, our findings suggest that TNFR1 may determine whether hUC-MSCs will gain abilities of anti-inflammation and tissue regeneration.

Keywords
Introduction
Rheumatoid arthritis (RA) is a chronic disease characterized by dysregulated immune cell activities and abnormal secretion of inflammatory cytokines1–3. Tumor necrosis factor α (TNF-α), produced by activated monocytes, macrophages, CD4+ T cells, and synovial fibroblasts, plays a crucial role in the pathogenesis of RA4,5. Initially, TNF-α and its receptors, tumor necrosis factor receptor 1 (TNFR1) and tumor necrosis factor receptor 2 (TNFR2), are produced as transmembrane proteins and are later released as soluble forms through cleavage by TNF-α–converting enzyme (TACE)6,7. TNF forward signaling occurs when either membrane or soluble TNF-α binds to transmembrane receptors, leading to the activation of inflammation. Conversely, reverse TNF signaling occurs when membrane or souble TNFR1 and TNFR2 combine with transmembrane TNF-α in immune cells, triggering the mitogen-activated protein kinase kinase 4 (MKK4) signaling pathway through the cytoplasmic domain of TNF-α. The reverse TNF signaling results in the production of transforming growth factor-β (TGF-β) and promotes monocyte apoptosis8,9. TNFR1 is expressed ubiquitously, while TNFR2 expression is restricted to Treg cells, endothelial cells, hematopoietic cells, and certain neuronal subpopulations10,11. Elevation of soluble TNFR1 or TNFR2 (sTNFR1/sTNFR2) has been observed in various diseases, including cancer, rheumatoid arthritis, neurological diseases, and kidney diseases12–15. These sTNFR1/sTNFR2 circulate in the human body and act as neutralizers of TNF-α to reduce inflammation.
The current approach to managing RA focuses on suppressing inflammation. However, the use of NSAIDs and conventional DMARDs, although effective in relieving inflammation and controlling the progression of RA, can have adverse effects, including kidney disease, heart attack, and gastrointestinal issues, which pose risks to patients 16 . To specifically target the key proinflammatory cytokines involved in RA development, various therapeutic options have been developed, including small molecule inhibitors, recombinant fusion proteins, and monoclonal antibodies16–19. Extensive research has been conducted on blocking the TNF-α/TNFR-axis to inhibit inflammation. Approved blockers include infliximab, etanercept, adalimumab, golimumab, and certolizumab pegol. It is worth noting that etanercept, a fusion protein consisting of extracellular TNFR2 and IgG1 Fc, demonstrates the feasibility of engineered soluble TNFR against TNF-α.
Mesenchymal stem cells (MSCs) have shown potential in the treatment of immune diseases, including RA, graft-versus-host disease (GvHD), and systemic lupus erythematosus (SLE), through modulating functions of immune cells and reducing inflammatory cytokine level20–30. In addition, their differentiation ability contributes to disease attenuation through the replacement of injured cells. For example, MSCs had been shown to differentiate into chondrocytes to repair damaged cartilage in osteoarthritis mice model 31 . However, the role of TNFR1 in promoting inflammation versus anti-inflammation remains unclear 32 . To elucidate the functions of TNFR1 in human umbilical cord–derived MSCs (hUC-MSCs)-mediated RA therapy, we assessed the performance of hUC-MSCs and siTNFR1-MSCs in inhibiting TNF-α production of PBMCs and alleviating RA symptoms in collagen-induced arthritis (CIA) mice. The secretion of growth factors and chemokines were investigated based on the results of animal experiments. These data suggest that hUC-MSCs will lose anti-inflammation ability and tissue recovery ability in the absence of TNFR1.
Materials and Methods
HUC-MSC Culture
Four hUC-MSCs cell lines were gifts from Beijing Baylx Biotech Co., Ltd. The cells were cultured in DMEM/F-12 (11320082, Thermo Scientific) supplemented with 10% FBS (97068-085, VWR) and bFGF (AF-100-18B, Peprotech) (final concentration 5 ng/ml) at 37°C and 5% CO2. Once the cells reached 90% confluency, they were passaged. The surface markers were assessed using FACS. Cells that showed ≥95% positive expression of CD73, CD90, and CD105, and ≤2% positive expression of CD19, CD34, CD31, CD11b, CD45, and HLA-DR were considered as standard hUC-MSCs. The standard cells were frozen at passage 2 (P2) for storage. For experiment use, cells were thawed and propagated. The TNFR1 was knocked down using commercial siTNFR1 as indicated (s14266, Ambion, validated) through lipofectamine RNAi-Max (13778150, Thermo Scientific). The MSCs used in animal experiments were never after P5.
PBMC Potency Assay
PBMCs was a gift from Beijing Baylx Biotech Co., Ltd. PBMCs were cultured in RPMI-1640 complete medium. For the potency assay, PBMCs were adjusted to a concentration of 1 × 107 cells/ml and then stimulated with anti-CD3 (317325, Biolegend) (final concentration: 1 μg/ml) and anti-CD28 (302933, Biolegend) (final concentration: 1 μg/ml) to induce TNF-α production. In 24-well plates, a total of 3 × 105 activated PBMCs were cocultured with 3 × 104 hUC-MSCs or siTNFR1-MSCs. The PBMCs alone were assigned to control. After 48 h of coculture, the conditioned medium was collected for measurement of TNF-α (DTA00D, R&D) and sTNFR1 (DRT100, R&D) using ELISA kits.
Mice Management
For efficacy evaluation of single dose of MSCs, male DBA/1J mice were purchased from Charles River Laboratories (Beijing, China). The mice were 6 weeks old upon receipt and were housed in a well-ventilated room with five mice per cage. The ambient temperature was maintained at 20°C–24°C, while the humidity was between 40% and 70%. The mice were exposed to 12 h of illumination daily (from 8:00 to 20:00) with an intensity of 15–20 lux. The feed and water were SPF grade and sterile. Prior to arthritis induction, mice were randomly divided into two groups: a healthy group consisting of five mice that did not receive any treatment, and a model group of 30 mice that were scheduled to receive two immunizations as shown in Fig. 3A. Collagen-induced arthritis (CIA) was constructed according to the protocol shared by Pietrosimone et al 33 . On the day of first immunization (day 0), each mice was injected 70 μl emulsion of Collagen II (CII) (20021, Chondrex) and Freund’s complete adjuvant (containing 1 mg/ml Mycobacterium tuberculosis) (CFA) (7001, Chondrex) at tail base through 23G/0.75 inch needle. On the day of boost immunization (day 21), mice were administered 100 µl emulsion of CII & Freund’s incomplete adjuvant (IFA) (7002, Chondrex) at the base of the tail. Three days after second immunization (day 24), each limb was double-blind rated on a scale of 0–4, and those mice with at least one leg having clinical score ≥2 were considered successfully modeled. Finally, CIA were successfully constructed in 23 mice and they were further randomly assigned to the control (8 mice), treatment I (8 mice), and treatment II (7 mice) groups. Meanwhile, one mouse in the healthy group died. Therefore, healthy group had four mice for observation. Treatment I and II were respectively given 2 × 106 hUC-MSCs and siTNFR1-MSCs (prepared by 80 nM siTNFR1 because 20 nM siTNFR1 had a quick restoration of TNFR1) suspended in multiple electrolyte injection solution (1707071404, Shijiazhuang NO.4 Pharmaceutical) at a concentration of 1 × 107 cells/ml. The control group was given 200 μl of carrier solution. Since the day before boost (day 21), paw thickness (0–10 mm calipers) and clinical score had been recorded every other day until the end of observation. On day 42, animals were euthanized by CO2 overdose and cervical dislocation. Their limbs were processed for histopathological analysis. Peripheral blood samples were collected on day 22 (200 μl), day 26 (200 μl), and at the end after euthanizing (1,000 μl). Clinical evaluation score: 0, no swelling; 1, mild redness and swelling of the tarsus or ankle joint; 2, mild redness and swelling from ankle to tarsus; 3, redness and swelling from ankle to metatarsal; 4, severe redness and swelling from ankle to toe, or joint stiffness 34 .
In addition, for hUC-MSCs duration in vivo, 25 7-week-old BALB/c mice were obtained from Vital River (Beijing, China) and were subjected to identical housing and environmental conditions as the DBA/1J males. They were given 2 × 106 hUC-MSCs at the tail vein. Mice were separately euthanized on day 0, day 1, day 3, day 5, and day 7 after injection, with five mice per timepoint. Day 0 refers to 6 h postinjection. After euthanizing, their eyeballs were quickly removed to collect 1 ml of blood for human TNFR1 and human HGF ELISA detection.
The animal study was approved by the animal ethics committee Laboratory Animal Center, Beijing Yizhuang Biomedical Park (Approval number 2019S035, Approval date June 4, 2019).
Histopathological Evaluation
After euthanizing, excessive fur and muscle were removed from animal limbs. Their limbs were fixed in 4% paraformaldehyde (PFA) (158127, Sigma) for 2 days and subsequently decalcified (decalcification solution, consisting of 14% EDTA, 0.2%–0.4% PFA in 1× PBS, pH=7.4) for 3 to 4 weeks. The decalcification solution was changed every 2 to 3 days. Following decalcification, all tissues underwent processing according to the 18-h protocol. The resulting 10-μm paraffin sections were stained with hematoxylin and eosin (H&E). H&E slices were observed upon 100× magnification.
Blood Cytokine Measurement
Collected peripheral blood was centrifuged at 7,500 r/min (Shanghai, China, Thermo, ST 16R) for 15 min to obtain serum. The IFN-γ, IL-6, and TNF-α in the serum were Luminex determined (Millipore systems). The resulting data are presented as the mean ± SEM.
Cytokines Secretion After Stimulation
The hUC-MSCs and siTNFR1-MSCs generated by 80 nM siTNFR1 were plated into 24-well plates (3 × 104 cells per well). After incubation overnight, recombinant human IFN-γ (285-IF, R&D systems) (10/20/100 ng/ml final concentration), IL-6 (10395-HNAE, Sino) (10/20/40 ng/ml final concentration), and TNF-α (GMP-10602-HNAE, Sino) (1/10/20 ng/ml final concentration), either alone or in combination, were added to the cells for a 24-h stimulation. After stimulation, the condition medium was harvested and centrifuged at 2,000 r/min for 10 min to get supernatant. The TNFR1 (DRT100, R&D systems), HGF (ab275901, Abcam), and KGF (ab183362, Abcam) in supernatant were measured through ELISA kits. The CXCL9, CXCL10, MCP-1, IL-8, and RANTES were measure through multiplex assay (MTH17MAG-47K, Millipore). The data are presented as mean ± SD.
TNFR1 Expression After Stimulation
hUC-MSCs were cultured in T25 flasks. When the cells reached 70%~80% confluency, they were stimulated with 100 ng/ml IFN-γ, 20 ng/ml TNF-α, or 40 ng/ml IL-6 for 24 h. After stimulation, the cells were washed with 1× PBS and lysed using RIPA lysis buffer (P0013B, Beyotime). The protein concentration was measured using BCA assay kits (P0011, Beyotime). Proteins were boiled at 95°C for 10 min and run on 10% SDS-PAGE gel. TNFR1 (1:500, 21574-1-AP, Proteintech) (1:500, PA5-81080, Thermo Scientific) and GAPDH (1:5,000, 2118, Cell Signaling) were detected at 4°C overnight.
Statistical Analysis
GraphPad Prism 9.0 and Excel were used to compute and graph the data. The normality was checked by Shapiro-Wilk test. To compare the difference between multiple treatments, we conducted One-way ANOVA followed by Dunnett’s comparisons. To compare the difference between treatments and control, we conducted One-way ANOVA followed by Tukey comparisons. The P-value < 0.05 was considered significant. Specifically, to compare paw thickness after Control, hUC-MSCs, and siTNFR1-MSCs treatment, we conducted two-way ANOVA followed by Bonferroni’s test, taking the effect of time into consideration.
Results
TNF-α Production in PBMCs Was Inhibited by hUC-MSCs in a TNFR1 Dose-Dependent Manner
The hUC-MSCs from four donors could secret sTNFR1 (Fig. 1A). They could inhibit the production of TNF-α in active PBMCs (Fig. 1B). This inhibition was partially impaired when PBMCs were cocultured with siTNFR1-MSCs in a dose-dependent manner (Fig. 1B). The inhibition of TNF-α production decreased from 89.0% (0 nM siTNFR1) to 45.6% (20 nM siTNFR1) (P < 0.01, Fig. 1C). Interestingly, although the secretion of sTNFR1 was similar between the 10 nM siTNFR1-MSC and 20 nM siTNFR1-MSC (Fig. 1A), the 10 nM siTNFR1-MSC led to stronger TNF-α inhibition, suggesting that cellular TNFR1 in MSCs may also contribute to the inhibition of TNF-α.

TNFR1 expression by hUC-MSCs contributes to the inhibition of TNF-α secretion. (A) sTNFR1 secretion after 24 h of siTNFR1 transfection. The data were analyzed using one-way ANOVA followed by Dunnett’s test. (B) TNF-α secretion after 48 h of coculture. The activated PBMCs were cultured with hUC-MSCs in the presence or absence of siTNFR1. The data were analyzed using one-way ANOVA followed by Tukey’s test. PP represents the PBMC alone group. (C) The TNF-α inhibition in response to siTNFR1 concentration. TNF-α Inhibition Rate = (1 – TNF-α secretion [hUC-MSC/PBMC] / TNF-α secretion [PBMC alone]) × 100%. Data are presented as mean ± SEM. The data were analyzed using one-way ANOVA followed by Tukey’s test. *P < 0.05 and **P < 0.01.
Function Check of hUC-MSCs
To investigate the impact of TNFR1 during hUC-MSC treatment in vivo, hUC-MSCs were expanded. Any cell line that showed altered activities after passages, such as abnormal proliferation, low transfection efficiency, or changes of cytokine secretion, was excluded from further experiments. Only one hUC-MSC cell line consistently secreted sTNFR1 after multiple passages (Fig. 2A) and therefore continued for further experiments. This cell line was intravenously injected in healthy BALB/c mice to observe the sTNFR1 secretion in vivo and estimate duration of hUC-MSC in mice. The sTNFR1 could be detected in the peripheral blood within 5 days after injection (Fig. 2B). In contrast, human KGF was still detectable on day 7 (Fig. 2C), suggesting that hUC-MSC could persist in BALB/c mice for 7 days and sTNFR1 secretion might be influenced by environment.

TNFR1 expression after several passages and TNFR1 expression duration in mice. (A) The secretion of sTNFR1 at different passages. siTNFR1-MSCs were transfected with 80 nM siTNFR1. (B and C) Secretion of sTNFR1 and KGF by hUC-MSCs after intravenous injection into BALB/c mice. Data are presented as mean ± SEM.
Mitigation of RA Symptoms by hUC-MSCs Was TNFR1-Dependent
To investigate the influence of TNFR1 during hUC-MSC treatment, we induced arthritis in DBA/1J mice using collagen II (CIA mice) and divided them into three groups: control (8 mice), treatment I (8 mice), and treatment II (7 mice), while four DBA/1J mice without CIA construction were assigned to healthy group. The treatment I group received 2 × 106 hUC-MSCs, while the II group received 2 × 106 siTNFR1-MSCs (Fig. 3A). The knockdown of TNFR1 was confirmed prior to the animal experiment (Fig. 2A). The observation lasted for 18 days.
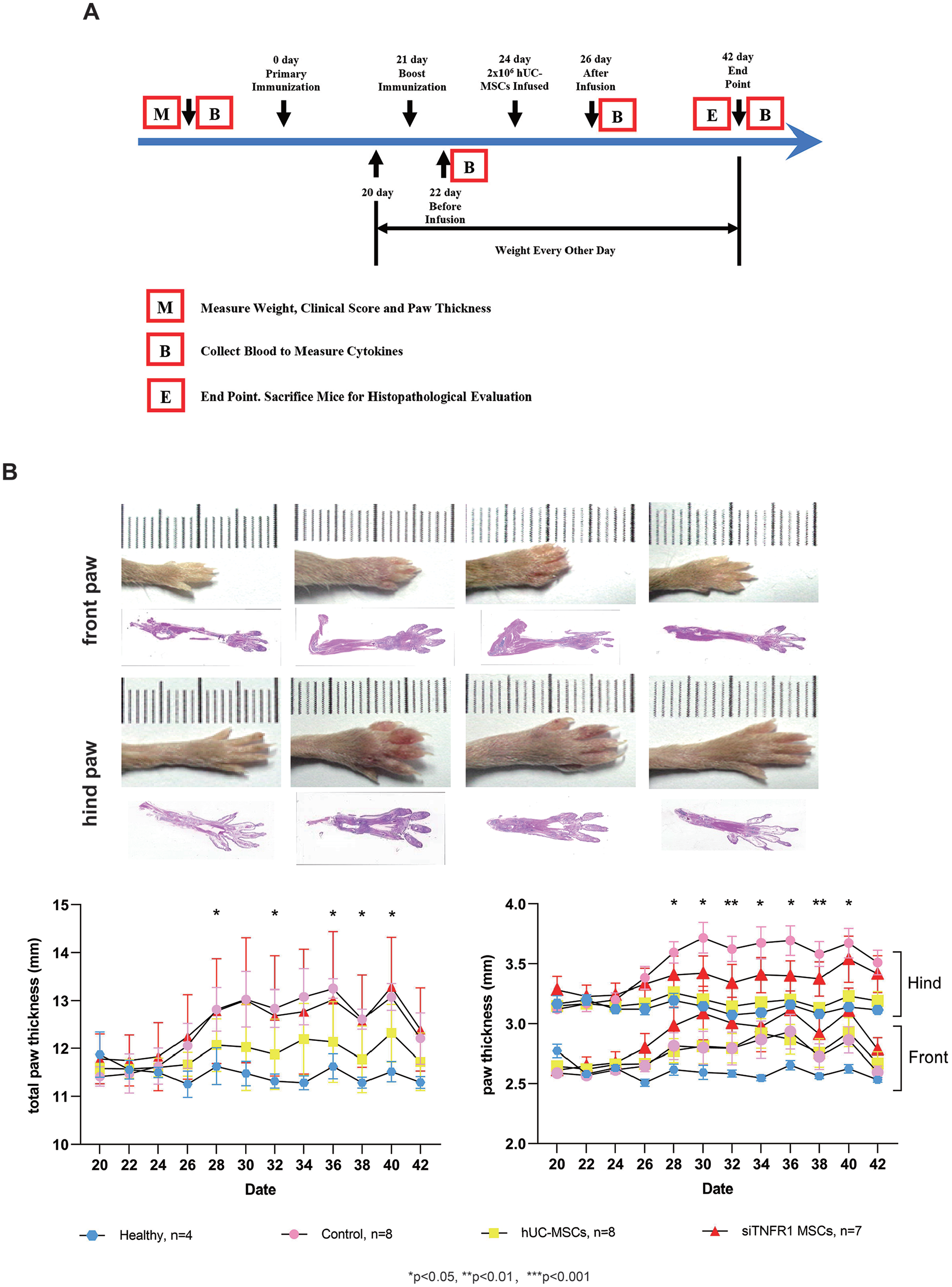
hUC-MSCs alleviated paw swelling in CIA mice. (A) Timeline of the animal experiment. (B) Changes in paw thickness after arthritis induction and therapy. Total paw thickness represents the combined measurement of front and hind paws. Data are presented as mean ± SEM. The data were analyzed using two-way ANOVA followed by Bonferroni’s test. *P < 0.05 and **P < 0.01.
Arthritis induction led to paw swelling, especially on hind paw. The control group exhibited the most severe swelling, as indicated by the total paw thickness and hind paw thickness (Fig. 3B). hUC-MSC alleviated the swelling, while siTNFR1-MSCs lost the healing effect (Fig. 3B). H&E staining of limbs revealed significantly differences in pathological status between groups. Compared to healthy mice, control mice exhibited a loss of bone architecture. Their joint cavity was invaded by synovial pannus. Cartilage damage was severe, as shown by the loss of superficial cartilage layer and subchondral bone erosion. In addition, severe inflammatory infiltration was founded in the limbs of control mice. The effect of siTNFR1-MSCs treatment was limited, as the bone architecture and other signs of arthritis were not fully alleviated. In contrast, the bone architecture after hUC-MSC was more similar to that of healthy mice, with significant recovery of cartilage layer and subchondral bone, although one area of erosion was still present. Furthermore, inflammatory infiltration was also suppressed.
The inflammatory cytokine level in mice peripheral blood was monitored. The inflammatory cytokines IL-6, TNF-α, and IFN-γ were significantly elevated after arthritis induction (Fig. 4B, control group). After hUC-MSC treatment, IL-6 significantly decreased on Day 26 and Day 42. hUC-MSC treatment reduced TNF-α level, but the significant difference was only observed on day 42 compared to the control. hUC-MSC treatment also reduced IFN-γ levels on Day 26 and Day 42, although a significant difference was not observed. In contrast, IL-6, TNF-α, and IFN-γ were not suppressed by siTNFR1-MSCs, indicating that TNFR1 was necessary for anti-inflammation of hUC-MSCs in vivo.
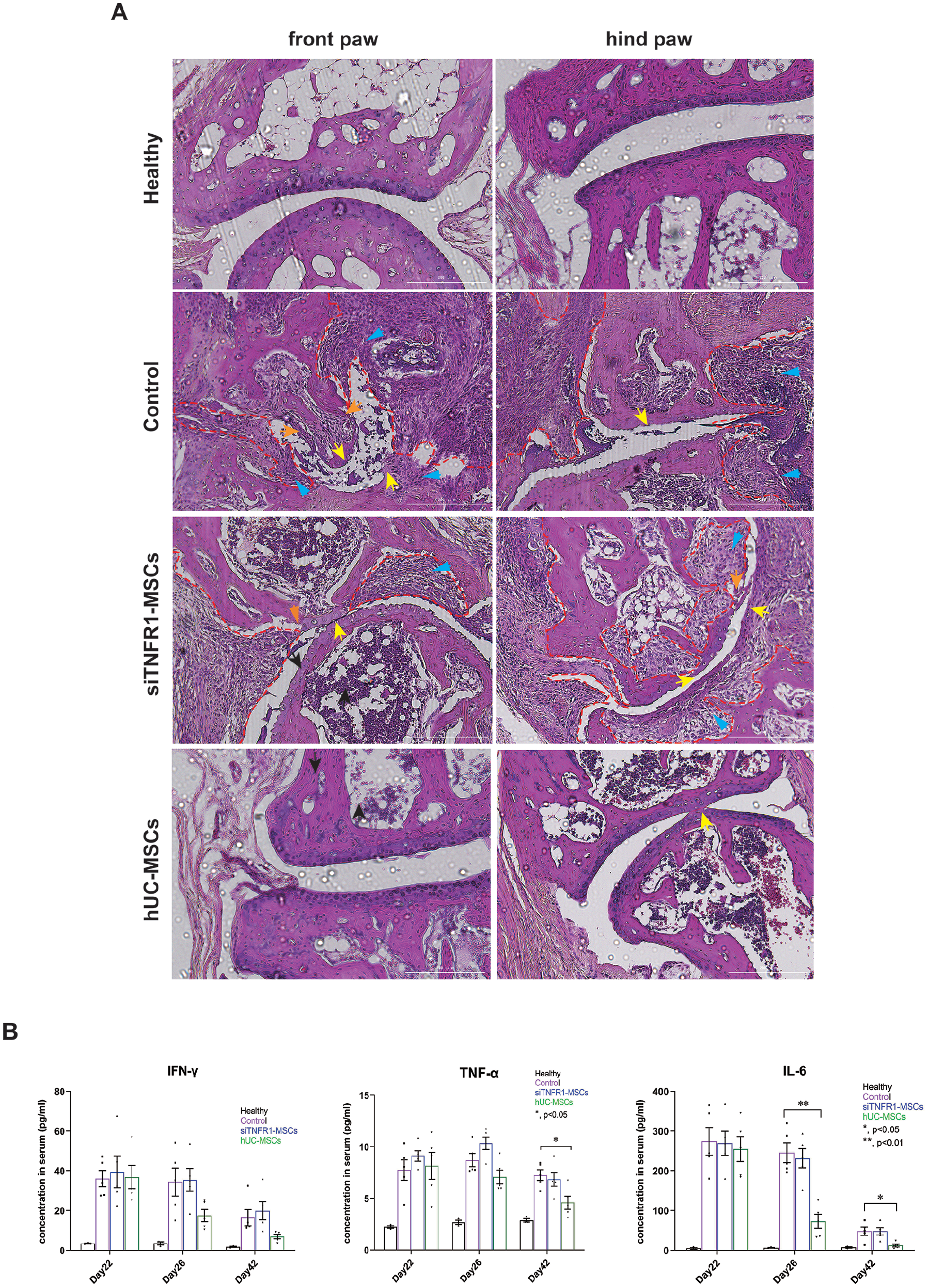
The amelioration of arthritis was impaired when TNFR1 expression was knocked down. (A) Representative H&E images of limbs. Synovial pannus invasion, red dash line; inflammatory infiltration, blue arrow; erosion of superficial cartilage layer, yellow arrow; subchondral bone erosion, organ arrow. White scale bar: 200 μm. (B) Inflammatory cytokines in the peripheral blood were measured before (day 22) and after cell therapy (day 26 and day 42). Data are presented as mean ± SEM. The data were analyzed using one-way ANOVA followed by Tukey’s test. *P < 0.05 and **P < 0.01.
During the observation period, no abnormalities were detected in the skin, behavior, excreta, diet, or injection sites of the mice. In addition, no mice died during the treatment. The only observed organ pathogenesis was tail biting in some control mice. In summary, the administration of hUC-MSCs did not result in any side effects.
TNFR1 Level Reduced in Response to Inflammatory Cytokines
We questioned whether TNFR1 expression would be upregulated in vivo in response to inflammatory environment. Unfortunately, due to the gradual death of hUC-MSCs after injection, the human sTNFR1 in peripheral blood could not accurately reflect real-time expression. To overcome this limitation, we conducted an in vitro experiment where hUC-MSCs were stimulated with gradient concentrations of IFN-γ, TNF-α, and IL-6 for 24 h. Subsequently, we analyzed the cellular TNFR1 and sTNFR1 levels. As illustrated in Fig. 5A, two antibodies targeting regions 233–455 (intracellular region) or 1–211 (extracellular region), respectively, recognized the full-length TNFR1 protein, which has an approximate molecular weight of 55 kDa (orange box). Interestingly, exposure to various cytokines, particularly TNF-α, resulted in a reduction of cellular TNFR1 levels. The antibody mapping region 233–455 not only detected the full-length TNFR1 but also identified a smaller fragment under 35 kDa (green box), which was possibly the proteolytic cleavage of TNFR1 mediated by TACE. Conversely, the antibody targeting region 1–211 did not detect any fragments. Given that TNFR1 was detected from whole cell lysates, this suggested that the cleavage of TNFR1 occurred on the surface of hUC-MSCs, as intracellular cleavage would yield detectable fragments containing the extracellular region. The extracellular region was released into medium and became sTNFR1. In addition, 1–211 antibody revealed an increase in full-length TNFR1 in siTNFR1-MSCs upon stimulation. When comparing unstimulated hUC-MSCs and siTNFR1-MSCs, the knockdown of TNFR1 in siTNFR1-MSCs indicated effective siTNFR1 functionality. Therefore, the observed upregulation of TNFR1 in response to stimulation may be attributed to the induction of TNFR1 mRNA or reduced TNFR1 degradation, rather than siTNFR1 malfunction. Consistent with cellular TNFR1, the secretion of sTNFR1 decreased after IFN-γ and TNF-α treatment (Fig. 5B). Furthermore, a slight decrease of sTNFR1 was observed after low concentration IL-6 stimulation, but this decrease was not observed after high concentration stimulation, which contradicted the findings for cellular TNFR1.

TNFR1 was reduced after 24 h stimulation with inflammatory cytokines. (A) Cellular TNFR1 level. Full-length blots are provided in Supplementary Fig. S1. (B) sTNFR1 level in growth medium. Data are presented as mean ± SD.
Loss of TNFR1 Impaired Production of Growth Factors and Chemokines
Considering the limited recovery of limbs after siTNFR1-MSCs treatment (Fig. 4A), we investigated whether TNFR1 also affected the tissue regeneration ability of hUC-MSCs. To address this, we compared the secretion of HGF and KGF (FGF-7) between hUC-MSCs and siTNFR1-MSCs in the presence or absence of combined inflammatory cytokine stimulation. HGF and KGF were selected because of their known involvement in MSC-mediated protection against lung and liver injuries35–38. We found that both HGF and KGF were significantly reduced after TNFR1 knockdown in the absence of stimulation (Fig. 6). Although stimulation led to a mildly decrease in HGF and KGF production in hUC-MSCs, this effect was not observed in siTNFR1-MSCs (Fig. 6). Furthermore, since hUC-MSCs can produce chemokines to regulate immune cells, to investigate whether this ability was impaired by TNFR1 knockdown, we compared the changes of various chemokines secretion under same conditions. Our results showed that hUC-MSCs greatly promoted CXCL9, CXCL10, and RANTES production after stimulation, while TNFR1 knockdown compromised their production (Fig. 6). TNFR1 knockdown led to stronger upregulation of MCP-1 after stimulation. In comparison, the upregulation of IL-8 was only observed in hUC-MSCs. These results suggested that TNFR1 knockdown did alter secretion of chemokines. To explore potential interaction between TNFR1 and these affected proteins, we did analysis upon STRING database (https://string-db.org). As shown in Fig. S2, functional and physical interactions were found between MCP-1 and CXCL9, CXCL10, RANTES, IL-8. However, experimentally verified interaction was not found between TNFR1 and growth factors or chemokines (Fig. S2.B). These suggest that the effect of TNFR1 was indirect.

Changes of cytokine secretion after 24 h of stimulation with inflammatory cytokines. Data are presented as mean ± SD.
Discussion
Soluble TNFR can inhibit TNF-α-mediated inflammatory pathways, making it a potential target for RA treatment. TNFR: Fc proteins, such as etanercept, have been developed for this purpose 39 . The four hUC-MSCs cell lines we obtained inhibited TNF-α production by PBMCs, which is consistent with previous research highlighting the anti-inflammatory abilities of MSCs40,41. Studies in mice have indicated that TNFR1 contributes to chronic inflammation, while TNFR2 exerts immune suppression41,42. However, there was reporting showing TNFR1 expressing MSCs inhibiting Th17 differentiation and alleviating encephalomyelitis in mice 43 . Our coculture displayed TNFR1 expression in hUC-MSCs contributed to inhibition of TNF-α production, suggesting TNFR1, at least in the case of hUC-MSCs, participating in immune suppression.
MSCs have limited capacity for proliferation as they undergo senescence after several rounds of propagation44,45. However, large-scale propagation is necessary for clinical use, and it is important to ensure that cytokine expression remains stable during this process. Among the different hUC-MSCs cell lines tested, only one demonstrated stable sTNFR1 level before P8. However, this cell line rapidly stopped secreting sTNFR1 after injection into BALB/c mice. The reduced secretion may be partially attributed to cell death and the absence of an inflammatory environment, as previous studies have shown that sTNFR1 production increases when MSCs are exposed to inflammatory stimulation43,46.
The therapeutic effect of hUC-MSCs and siTNFR1-MSCs was evaluated in DBA/1J mice. hUC-MSCs injection significantly improved pathological conditions and reduced proinflammatory cytokine levels. However, siTNFR1-MSCs had lesser effects in this regard. The comparison of cytokine levels on Day 26 and Day 42 revealed that IL-6 and IFN-γ, rather than TNF-α, were the most affected by TNFR1 knockdown. These results suggest that TNFR1 plays a crucial role in determining whether hUC-MSCs have anti-inflammation and regeneration abilities in vivo. According to findings of Waetzig GH et al. and Yagi H et al.9,46, sTNFR1 may contribute partially to the anti-inflammatory response. It was observed that the secretion of sTNFR1 increased when MSCs were exposed to inflammatory serum after 24 h43,46. However, our cytokine stimulation experiment could not reproduce this phenomenon, but it revealed altered secretion of CXCL9, CXCL10, RANTES, IL-8, and MCP-1 after TNFR1 knockdown. These chemokines participate in migration of immune cells, such as monocytes, basophiles, T cells, eosinophils, neutrophiles. In human cells, the production of these chemokines could be regulated in response to IFN-γ, TNF-α, IL-6, either individually or in combination, with involvement of the JAK/STAT, NF-КB, or PI3K-AKT pathways47–55. Involvement of TNFR1 in these signaling pathways has been verified in various cell types. For example, TNFR1 and Jak1 exhibit a rapid and transient interaction in human primary B cells upon TNF-α stimulation 56 . However, under different condition, TNFR1 may act like a double-edged sword. It could induce neutrophil apoptosis through recruitment of TRADD upon TNF-α stimulation, with activation of p38 and PI3K 57 . Conversely, TNFR1 promoted survival of CD8+ T cells by interacting with TRAF2 to activate NF-κB 58 . According to Tergaonkar et al., the status of RIPK1, caspase-8, and MLKL determine whether TNFR1 form complex I, IIa, IIb, which initiates NF-κB/MAPK, caspase, RIPK3-MLKL, respectively, driving cell fate toward survival/proinflammation, apoptosis, or necrosis 59 . This may explain why MSCs display anti-inflammation and therapeutic effects after inflammatory cytokines priming. Trigueros et al. demonstrated that NF-КB p65 translocated to nucleus of human bone marrow–derived MSCs (hBM-MSCs) in response to TNF-α, and that hBM-MSCs-mediated inhibition of T-cell proliferation required NF-КB, both of which were TNFR1-dependent 28 . Furthermore, Chen et al. demonstrated that RIPK1 was constitutively expressed in hBM-MSCs 60 . Therefore, the activation of NF-КB may be driven by posttranslational modifications of RIPK1 following the assembly of TNFR1-initiated complex in MSCs. In our study, siTNFR1-MSCs likely had lost the TNFR1 complex and the possibility of interacting with signaling pathway component, such as Jak1, thus influenced chemokine production and impaired the ability to regulate migration of immune cells, leading to the strong inflammation infiltration in siTNFR1-MSCs-treated animals.
TNFR1 knockdown reduced HGF and KGF production through unknown mechanisms. HGF is initially produced as pre-pro-HGF and then undergoes cleavage between Arg494 and Val495 to form pro-HGF 61 . The pro-HGF is secreted out of cells and activated in serum through cleavage at the Arg-Val site. Various proteases, such as HGF activator, urokinase-type plasminogen activator, and metalloproteinases, can activate HGF in serum 61 . The mature HGF binds to its receptor c-MET, a transmembrane protein, and activates several signaling pathways, including PI3K/AKT, STAT3, MAPK, ERK-1/2, and b-catenin/Wnt 62 . These signaling pathways contribute to cell proliferation, resistance to apoptosis, and angiogenesis. The HGF/c-MET pathway is necessary for liver regeneration and repair 63 . HGF secreted by MSCs has been shown to regulate T cells differentiation and promote regeneration in spinal cord injury, lung injury, and many other immune-related diseases36,64–66. In our hUC-MSCs, TNFR1 may directly or indirectly affect extracellular HGF level by promoting expression of HGF or HGF processors. The secreted HGF may bind to the c-MET in mice tissue to induce limb recovery. Besides, the HGF secreted by hUC-MSCs themselves may be essential for their migration ability, as shown by the effect of HGF on mouse MSCs revealed by Forte G et al 67 . KGF, however, has been identified as one of the effective components in MSCs’ secretome for therapy68,69. KGF binds to its receptor FGFR in the presence of cofactors heparin sulfate (HS) and heparin proteoglycans, leading to formation of FGF-FGFR-HS complex. The intracellular tyrosine kinase domain of FGFR is then activated and induces signaling pathways including MAPK, PI3K-AKT, PLCγ, and STAT, which is important for tissue repair, regeneration, and metabolism 70 . It has been shown that KGF expressed by MSCs may protect AT-II cells from injury induced by inflammation through PI3K/Akt/mTOR signaling pathway 71 . The previous findings indicate that KGF mediated pathways may also partially promote limb recovery during hUC-MSCs treatment.
This study has several shortcomings. First, the subpopulation of immune cells in peripheral blood and lesions, such as T-cells, should be evaluated. Second, the distribution of hUC-MSCs to the lesions needs to be assessed. Third, comparing to paw thickness, measuring paw volume would be better. Throughout the study, no side effects of hUC-MSCs were observed. Currently, UC-MSCs have entered clinical trials for RA therapy72,73. A study demonstrated safety of UC-MSCs when they were in combination with DMARDs, and maintained alleviation of RA symptoms 3 years after treatment 74 . However, a challenge of this combination is that DMARDs may interfere with the biological effects of MSCs. As revealed by Mogi et al., the adipogenesis, osteogenesis, and chondrogenesis of human cartilage–derived primary MSCs was inhibited by DMARDs, such as methotrexate, in a dose-dependent way in vitro 75 . For in vivo assessment of MSCs, it is worthy to include a combination of MSCs with other type of RA therapy agents to determine whether the specific function of MSCs, such as tissue regeneration, or differentiation, will be influenced.
Conclusion
hUC-MSCs inhibited inflammation in vitro and in vivo. This anti-inflammation ability required TNFR1. Loss of TNFR1 influenced production of growth factors and chemokines. However, the pathways that link TNFR1 to growth factors or chemokines are not clear.
Supplemental Material
sj-jpg-1-cll-10.1177_09636897241301703 – Supplemental material for Tumor Necrosis Factor Receptor 1 Is Required for Human Umbilical Cord–Derived Mesenchymal Stem Cell–Mediated Rheumatoid Arthritis Therapy
Supplemental material, sj-jpg-1-cll-10.1177_09636897241301703 for Tumor Necrosis Factor Receptor 1 Is Required for Human Umbilical Cord–Derived Mesenchymal Stem Cell–Mediated Rheumatoid Arthritis Therapy by Guangyang Liu, Herui Wang, Chenliang Zhang, Xin Li, Yi Mi, Yaoyao Chen, Liqiang Xu, Li Miao, Haomiao Long and Yongjun Liu in Cell Transplantation
Supplemental Material
sj-jpg-2-cll-10.1177_09636897241301703 – Supplemental material for Tumor Necrosis Factor Receptor 1 Is Required for Human Umbilical Cord–Derived Mesenchymal Stem Cell–Mediated Rheumatoid Arthritis Therapy
Supplemental material, sj-jpg-2-cll-10.1177_09636897241301703 for Tumor Necrosis Factor Receptor 1 Is Required for Human Umbilical Cord–Derived Mesenchymal Stem Cell–Mediated Rheumatoid Arthritis Therapy by Guangyang Liu, Herui Wang, Chenliang Zhang, Xin Li, Yi Mi, Yaoyao Chen, Liqiang Xu, Li Miao, Haomiao Long and Yongjun Liu in Cell Transplantation
Footnotes
Acknowledgements
We acknowledge citexs (![]() ) for providing the service of language editing. We appreciate MD. Qinggang Ge (Department of Critical Care Medicine, Peking University Third Hospital, Beijing, China) supervised the use of human cells in this study. The graphical abstract was created by MedPeer. We appreciate their platform of scientific image.
) for providing the service of language editing. We appreciate MD. Qinggang Ge (Department of Critical Care Medicine, Peking University Third Hospital, Beijing, China) supervised the use of human cells in this study. The graphical abstract was created by MedPeer. We appreciate their platform of scientific image.
Author Contributions
G.Y.L.—Conceptualization, Supervision, Funding acquisition, Formal analysis, Writing-review & editing; H.R.W.—Methodology, Investigation, Formal analysis, Writing-original draft; C.L.Z.—Data curation, Project administration, Resources; X.L., Y.M., L.Q.X., and L.M.—Investigation; Y.Y.C.—Project administration; H.M.L.—Resources; Y.J.L.—Conceptualization, Supervision, Funding acquisition. All authors read and approved the final manuscript.
Availability of Data and Material
The data used during in this study are available from the corresponding author on reasonable request.
Ethical Approval
The animal study was approved by the animal ethics committee Laboratory Animal Center, Beijing Yizhuang Biomedical Park (Approval number 2019S035, Approval date June 4, 2019). The use of human umbilical cord–derived cells and peripheral blood mononuclear cells was approved by Peking University Third Hospital Medical Science Research Ethics Committee (Approval number M2023825).
Statement of Human and Animal Rights
This article does not contain any studies with human or animal subjects.
Statement of Informed Consent
There are no human subjects in this article and informed consent is not applicable.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by grants from the Beijing Municipal Science and Technology Project (Nos Z221100007922028 and Z211100002521006). The funding body played no role in the design of the study and collection, analysis, and interpretation of data and in writing the manuscript.
Supplemental Material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.