
Abstract
Pulmonary fibrosis is a complex and multifactorial condition that involves a cascade of events, including lung injury, damage of alveolar epithelial cells (AECs), generation of immune cell–driven inflammation, and activation of fibroblasts and their differentiation into myofibroblasts, resulting in the excessive production and deposition of collagen and progressive scarring and fibrosis of the lung tissue. As lung fibrosis advances, the scarring and stiffening of lung tissue can significantly hinder the exchange of oxygen and carbon dioxide, potentially leading to respiratory failure that can be life-threatening. Anti-inflammatory and immunosuppressive drugs are used to slow down the progression of the disease, manage symptoms, and enhance the patient’s quality of life. However, prolonged immunosuppression could increase the susceptibility to severe bacterial, viral, or fungal pneumonia in lung-transplant recipients. Therefore, there is an urgent need for new therapeutic agents that can effectively reduce lung inflammation and fibrosis without compromising the protective immune response in patients with severe lung fibrosis. Results obtained in recently published studies demonstrated that mesenchymal stem/stromal cell-derived microRNAs (MSC-miRNAs) could attenuate detrimental immune response in injured lungs and prevent progression of lung fibrosis. Through the post-transcriptional regulation of target mRNA, MSC-miRNAs modulate protein synthesis and affect viability, proliferation, and cytokine production in AECs, fibroblasts, and lung-infiltrated immune cells. In order to delineate molecular mechanisms responsible for beneficial effects of MSC-miRNAs in the treatment of lung fibrosis, in this review article, we summarized current knowledge related to anti-fibrotic and anti-inflammatory pathways elicited in immune cells, AECs, and myofibroblasts by MSC-miRNAs.
Pulmonary Fibrosis and Current Therapies
Pulmonary fibrosis is a progressive disease characterized by the excessive and abnormal accumulation of fibrous or connective tissue in the lungs 1 . It is a complex and multifactorial condition that involves a cascade of events, starting with lung injury, followed by inflammation, activation of fibroblasts, excessive collagen deposition, and progressive scarring of the lung tissue1,2. The continual and repeated injuries of alveolar epithelial (AECs) and endothelial cells (ECs) and subsequent destruction of the alveolar–capillary basement membrane, caused by microbial pathogens, irradiation, drugs, and environmental pollutants and hazards, represent an initial step in the pathogenesis of lung fibrosis 1 . Alarmins and damage-associated molecular patterns (DAMPs), which are massively released from injured AECs and ECs, bind to their receptors on alveolar macrophages and dendritic cells (DCs) and activate several pro-inflammatory signaling pathways 3 . Upon binding to toll-like receptors (TLR)-2, TLR-4, and TLR-9, high mobility group box 1 (HMGB1), heat shock proteins (HSPs), and adenosine triphosphate (ATP) activate MyD88 and TRIF-driven signaling, which results in increased production of inflammatory cytokines (tumor necrosis factor-alpha [TNF-α], interleukin-1 beta [IL-1β], interferon gamma [IFN-γ]). Viral RNAs or self-RNA released from damaged cells are being recognized by retinoic acid-inducible gene I-like receptor (RLR), including retinoic acid-inducible gene I (RIG-I) and melanoma differentiation-associated gene 5 (MDA5) 3 . Activation of RLRs leads to the recruitment of the mitochondrial antiviral-signaling protein (MAVS) and the subsequent activation of transcription factors, such as NF-κB and IRF3/7 1 , 3 . This results in the induction of IFN-driven inflammatory response in the lungs. Similarly, carbohydrate-containing antigens of microbial pathogens bind to C-type lectin receptors (CLRs) on alveolar macrophages2,3. This interaction induces assembly and activation of the CARD9-BCL10-MALT1 (CBM) complex, which induces activation of NF-κB, resulting in the increased production of pro-inflammatory cytokines IL-1β, IL-6, and TNF-α 3 . Mitochondrial DNA, released from injured AECs and ECs, induces generation of NLRP3 inflammasome in lung neutrophils and alveolar macrophages 3 . NLRP3 inflammasome activates caspase-1, enabling processing and secretion of IL-1β and IL-18. Under the influence of inflammatory TNF-α and IL-1β, E and P selectins are being increasingly expressed on ECs, enabling massive recruitment of circulating leukocytes in injured lungs 3 . As a result, the lungs become infiltrated with inflammatory, nitric oxide (NO), and IL-6-producing macrophages and neutrophils; IFN-γ and IL-17-producing CD4+ Th1 and Th17 lymphocytes; and cytotoxic immune cells (natural killer [NK], NK T cells, CD8+ T lymphocytes [CTLs], which in TNF-α, FasL, and TRAIL-dependent manner induce apoptosis of AECs). CD4+ Th1 cells in IFN-γ and CD4+ Th17 cells in an IL-17-dependent manner generate inflammatory phenotype in macrophages and neutrophils, enabling the creation of “positive loop,” which aggravates on-going lung inflammation1,3.
Continuously elevated levels of inflammatory cytokines activate pro-fibrotic transcriptional factors (the proviral integration site for Moloney murine leukemia virus 1 [PIM1] and nuclear factor of activated T cells-1 [NFATc1]) in fibroblasts, inducing enhanced expression of genes that are responsible for collagen, elastin, and fibronectin formation3,4. In addition, activated fibroblasts differentiate into myofibroblasts 4 . This activation is often triggered by pro-fibrotic factors transforming growth factor-beta (TGF-β) and platelet-derived growth factor (PDGF), which are released by activated, lung-infiltrated macrophages 3 . TGF-β is a key regulator of myofibroblast differentiation and collagen synthesis5,6. TGF-β binds to its receptors, which leads to the phosphorylation and activation of downstream effector proteins called Smads 5 . Once activated, Smads translocate into the nucleus and regulate the expression of target genes associated with myofibroblast differentiation and collagen, fibronectin, and alpha-smooth muscle actin (α-SMA) production 5 . In this way, TGF-β enhances the contractile properties of myofibroblasts, impairing lung function and reducing lung compliance5,6. TGF-β also stimulates the production of other pro-fibrotic cytokines and growth factors (IL-13, PDGF, connective tissue growth factor [CTGF], fibroblast growth factor [FGF]) amplifying the fibrotic response and promoting tissue remodeling5,6.
Pro-fibrotic and pro-inflammatory cytokines and growth factors released by inflammatory cells, activated fibroblasts, and myofibroblasts stimulate epithelial-to-mesenchymal transition (EMT) in the remaining healthy epithelial cells within the lung tissue 7 . EMT is a biological process where epithelial cells lose their characteristic features and acquire mesenchymal-like properties 8 . EMT-mediated damage and transformation of AECs leads to impaired gas exchange, crucially contributing to the progression of lung fibrosis7,8. In addition, EMT process allows the epithelial cells to migrate and invade the underlying lung tissue7,8. This migration and invasion contribute to the excessive accumulation of fibroblasts and myofibroblasts which, through the production of collagen and fibronectin, disrupt the normal architecture of the lung tissue, impairing its function 7 . The excessive collagen deposition and scar tissue formation continue to progress over time, leading to the progression of lung fibrosis7,8. This results in the thickening and stiffening of the lung tissue, making it increasingly difficult for the lungs to expand and contract during breathing 8 . As lung fibrosis worsens, lungs become less elastic and unable to optimally transfer oxygen from the air into the bloodstream 8 .
Difficulty in breathing, a dry cough, fatigue, chest pain, and discomfort are the characteristic symptoms of lung fibrosis, which may occur due to inflammation and scarring in the lung tissue 9 . However, the main clinical symptoms and signs of lung fibrosis can differ based on the stage and seriousness of the disease9,10. Some patients may experience rapid and shallow breathing (tachypnea) as they struggle to obtain sufficient oxygen 10 . As lung fibrosis advances, the scarring and stiffening of lung tissue can significantly hinder the exchange of oxygen and carbon dioxide, potentially leading to respiratory failure that can be life-threatening 10 .
The goal of treating lung fibrosis is to slow down the progression of the disease, manage symptoms, and enhance the patient’s quality of life11,12. To achieve this, immunomodulators such as corticosteroids and anti-fibrotic drugs with potential anti-inflammatory actions such as pirfenidone and nintedanib are usually prescribed12–15. These medications work to inhibit harmful immune responses, reduce inflammation, and slow down the development of fibrosis in the lungs. In cases where oxygen levels in the blood are low, supplemental oxygen therapy is often recommended12,14,15. For individuals with advanced lung fibrosis and severe respiratory impairment, lung transplantation may be the only viable treatment option12,13. However, the availability of suitable donor lungs and the need for lifelong immunosuppressive therapy pose challenges to this approach 12 . Despite the use of immunosuppressant drugs, there is still a risk of the immune system rejecting the transplanted lung12,13. In addition, prolonged use of immunosuppressants increases the susceptibility to severe bacterial, viral, or fungal pneumonia in lung transplant recipients 12 . Therefore, there is an urgent need for new therapeutic agents that can effectively reduce lung inflammation and fibrosis without compromising the protective immune response in patients with severe lung fibrosis 13 .
Results obtained in recently published experimental studies demonstrated that mesenchymal stem/stromal cell (MSC)-derived extracellular vesicles (MSC-EVs), due to their capacity to attenuate detrimental immune response in injured lungs, could be used as potentially new therapeutic agents in the treatment of lung fibrosis16–19. MSC-EVs represent heterogeneous population of MSC-sourced, nano-sized particles, which are, based on their size and biogenesis, classified into exosomes (MSC-Exos), microvesicles, and apoptotic bodies20,21. MSC-Exos are the smallest EVs (30–150 nm) formed from the inward budding of the endosomal membrane. MSC-derived microvesicles are larger in size (100–1,000 nm) and originate from direct budding off the plasma membrane 19 . Apoptotic bodies represent the largest type (up to 5,000 nm) of MSC-EVs generated during programmed cell death of MSCs and contain their cellular debris, including fragmented organelles and nucleic acids 19 . MSC-EVs serve distinct functions in cellular communication, immune responses, and tissue homeostasis, contributing to various physiological and pathological processes18–22. Each type of MSC-EVs contains a variety of MSC-sourced immunoregulatory and angiomodulatory proteins, lipids, DNA, and RNA (messenger RNA [mRNA], microRNA [miRNA], small interfering RNA [siRNA], transfer RNA, ribosomal RNA [rRNA], long non-coding RNA [lncRNA]) which may modulate the functional properties of neighboring and distinct cells that take them up during paracrine or endocrine communication with MSCs 22 . Importantly, MSC-EVs are able to safeguard RNAs and other MSC-sourced biomolecules from degradation while delivering them to lung-infiltrated immune cells, AECs, and myofibroblasts, affecting their viability, proliferation, and cytokine production19–22.
During the last few years, several research groups provided evidence that MSC-sourced miRNAs play crucially important role in MSC-EV–based attenuation of lung fibrosis23–25. Nataliya and colleagues 23 demonstrated that human adipose tissue–derived MSC-EVs (hAT-MSC-EVs), by delivering miR-29c and miR-129 in myofibroblasts, modulated the expression of vinculin, α-actinin 2, and α-actinin 4; inhibited the expression of α-SMA and its incorporation into stress fibers; and caused dedifferentiation of myofibroblasts which resulted in the resolution of on-going fibrosis in experimental mice. Zhou and coworkers 24 showed that human bone marrow–derived MSC-EVs (hBM-MSC-EVs) suppressed lung fibroblast activation and delayed progression of pulmonary fibrosis in mice by down-regulating the expression of SOX4 and DKK1 in miR186-dependent manner, while Lei et al. 25 demonstrated that human placenta–derived MSC-EVs (hPL-MSC-EVs) reduced the levels of radiation-induced DNA damage and attenuated lung vascular damage, inflammation, and fibrosis through transferring miR-214-3.
Although these findings strongly suggest therapeutic potential of MSC-derived miR-29c, miR-129, miR186, and miR-214-3 in the treatment of pulmonary fibrosis, results obtained in several recently published studies indicated that other miRNAs (miR-21, miR-155, miR-410, miR-221, mR-222, miR-9, miR-124) had a pathogenic role in the development and progression of lung fibrosis26–29. MiR-21, which is significantly up-regulated in fibrotic lungs, suppresses the expression of SPRY1 gene which is a negative regulator of the pro-fibrotic Wnt/β-catenin signaling pathway26,28. Elevated miR-155 levels lead to reduced expression of SOCS1 and SHIP1 genes that suppress production of pro-inflammatory and pro-fibrotic cytokine in lung-infiltrated immune cells, contributing to the development and progression of uncontrolled inflammation and fibrosis in the lungs27,28. MiR-221, miR-222, and miR-410 promote pulmonary fibrosis by enhancing proliferation of fibroblasts through the decreased expression of p27Kip1 and PTEN genes which regulate cell cycle arrest and apoptosis in the fibrotic lungs28,29. By modulating the expression of the ANO1 gene, MiR-9 promotes TGF-β/Smad-driven pulmonary fibrosis, while pro-fibrotic activity of miR-124 is relied on its capacity to regulate AXIN1-dependent activation of Wnt/β-catenin signaling in myofibroblasts 29 .
Since different miRNAs regulate various cellular processes involved in the development, progression, and attenuation of lung fibrosis, better understanding of their specific roles and mechanisms is critical for developing effective therapeutic strategies for the treatment of this disease 29 . Therefore, in order to delineate molecular mechanisms responsible for beneficial effects of MSC-miRNAs in the treatment of pulmonary fibrosis, in this review article, we summarized current knowledge related to anti-fibrotic, anti-inflammatory, and immunoregulatory pathways elicited in immune cells, AECs, and myofibroblasts by MSC-miRNAs. An extensive literature review was carried out in February 2024 across several databases (MEDLINE, EMBASE, Google Scholar), from 2000 to present. Keywords used in the selection were “mesenchymal stem/stromal cells,” “miRNAs,” “exosomes,” “lung fibrosis,” “lung inflammation,” “signaling pathways,” “immune cells,” “fibroblasts,” “myofibroblasts,” “immunoregulation,” “tissue repair and regeneration.” A comprehensive search was conducted, resulting in 102 articles. Two of the authors (CRH and VV) independently reviewed the abstracts of these articles to determine their relevance to the topic of this article. Only studies that focused on the molecular and cellular mechanisms involved in the modulation of lung fibrosis using MSC-miRNAs were considered eligible. The findings of these relevant studies were then analyzed and included in this review.
MSC: An Inexhaustible Cell Source of Immunoregulatory and Anti-Fibrotic miRNAs
MSCs are self-renewable, immunomodulatory stem cells that reside in almost all postnatal tissue and organs, including lungs30,31. In response to the injury of AECs and ECs, MSCs produce various growth factors and bioactive molecules that inhibit on-going lung inflammation and fibrosis 32 . MSC-sourced IL-1 receptor antagonist and soluble TNF receptors inhibit pro-inflammatory effects of IL-1β and TNF-α, down-regulate the expression of selectins and integrins on ECs, and prevent influx of circulating leukocytes in inflamed lungs30–33. MSC-derived Indoleamine 2, 3 dioxygenase (IDO) and IL-10 suppress the expression of co-stimulatory molecules and attenuated the production of inflammatory cytokines (IL-12, IL-4, IL-1β, IL-6, and IL-23) in lung-infiltrated DCs, preventing DC-dependent generation of Th1, Th2, and Th17 cell-driven immune response32,34. MSC-sourced IL-10 and prostaglandin E2 (PGE2) suppress synthesis and release of cytotoxic molecules (perforins and granzymes) from CTLs and enhance the expression of anti-apoptotic genes in AECs, attenuating CTL-dependent apoptosis of AECs in inflamed lungs32,34,35. In addition, MSC-derived hepatocyte growth factor (HGF), PGE2, and IL-10 act synergistically to inhibit synthesis of matrix metalloproteinases (MMPs) in alveolar macrophages and to suppress activation of pro-fibrotic transcriptional factors and synthesis of collagen in fibroblasts and myofibroblasts, efficiently preventing tissue remodeling and fibrosis32–36. Precisely, MSC-sourced HGF binds to a c-MET receptor on activated fibroblasts, inducing activation of PI3K/Akt and Ras/MAPK pathways, which lead to the inhibition of fibroblast activation and the subsequent reduction in collagen synthesis32,36. MSC-derived PGE2, HGF, and IL-10 interfere with TGF-β signaling by inhibiting the phosphorylation and nuclear translocation of Smad proteins, thereby inhibiting the transcription of pro-fibrotic collagen and α-SMA genes32,36. Moreover, MSC-derived secretome containing PGE2, IL-10, and HGF can also prevent the differentiation of fibroblasts into myofibroblasts, preventing the development of lung fibrosis30,32. Several lines of evidence suggested that among various immunoregulatory factors presented in MSC-sourced secretome, HGF was mainly responsible for beneficial effects of MSCs in the suppression of lung fibrosis36–38. BM-MSCs, in HGF-dependent manner, inhibited apoptosis and enhanced proliferation of AECs 37 . Human umbilical cord–derived MSCs (hUC-MSC) that over-expressed HGF (HGF-UCMSCs) completely suppressed bleomycin-induced lung fibrosis, remarkably improved respiratory function, and significantly attenuated IL-17–driven inflammation in experimental rats 38 . Similarly, when the HGF gene was knocked down in BM-MSCs, their capacity to suppress proliferation of fibroblasts and to inhibit bleomycin-induced lung fibrosis was completely abrogated, indicating that HGF is essential for effective MSC-based alleviation of pulmonary fibrosis 37 .
Finally, in addition to immunoregulatory proteins, MSCs also release pro-angiogenic factors (vascular endothelial growth factor [VEGF], IL-6, angipoietin), which improve blood vessel formation and oxygenation in fibrotic lungs32,39. Therefore, the cross-talk between MSCs, lung-infiltrated immune cells, AECs, and fibroblasts is crucially responsible for the regulation and maintenance of homeostasis in injured lungs and for the suppression of immune cell–driven lung inflammation and fibrosis 36 .
Although MSCs importantly contribute to the restoration of respiratory function in fibrotic lungs, several side effects caused by engrafted MSCs significantly limit their potential clinical use in the treatment of pulmonary fibrosis 40 . When MSCs from a different donor are transplanted, major histocompatibility (MHC) molecules on their membrane can trigger an allogeneic immune response and may elicit immune cell-driven inflammation 41 . Rarely, transplanted MSCs may spontaneously differentiate into unwanted cell types, particularly chondrocytes or osteocytes, significantly compromising the structure, integrity, and function of the injured tissues 40 .
To overcome these limitations, a large number of experimental studies focused their attention on the therapeutic application of cell-free, MSC-derived bioactive product: MSC-Exos 41 . MSC-Exos are nano-scaled spherical EVs that are capable to deliver MSC-sourced bioactive molecules directly in the recipient cells, modulating their phenotype and function42,43. MSC-Exos contain various MSC-derived growth factors (VEGF, PDGF) and anti-inflammatory proteins (IDO, IL-1Ra, PGE2, IL-10) which can affect survival, differentiation, and cytokine production in fibroblasts and immune cells that play pathogenic role in the development and progression of lung fibrosis41–43. Results obtained in large number of recently published experimental studies demonstrated beneficial effects of MSC-Exos in the treatment of lung fibrosis, suggesting their potential therapeutic use in clinical settings44–46. By delivering HSP-70, which has cytoprotective properties, MSC-Exos down-regulated the expression of pro-apoptotic genes and activated phosphatidylinositol-3-kinase (PI3K)/Akt-driven anti-apoptotic signaling pathway in AECs, improving their survival and viability44,45. MSC-Exos created an immunosuppressive environment in fibrotic lungs by inducing the expansion of T regulatory cells and alternatively activated (M2) macrophages46,47. MSC-Exo-containing IDO down-regulated the expression of genes that regulate Th17 differentiation and favored the activity of Treg-related FoxP3 transcriptional factor 42 . In mice model of pulmonary fibrosis, MSC-Exos, in IDO-dependent manner, induced the expansion of Tregs in the inflamed lungs and prevented their trans-differentiation into inflammatory Th17 cells, which led to the attenuation of ongoing inflammation46,47. MSC-Exos contained within derived-multiple allogeneic proteins paracrine signaling (d-MAPPS) inhalation solution suppressed Th17 cell–driven lung inflammation in experimental animals, inhibited production of pro-inflammatory and pro-fibrotic cytokines (IL-17, TNF-α, IL-1β), and enhanced secretion of anti-inflammatory IL-10 in human leukocytes48,49. MSC-Exos, in IL-10- and PGE2-dependent manner, generated an anti-inflammatory and immunosuppressive (M2) phenotype in alveolar macrophages. MSC-Exo-primed M2 macrophages increasingly produced immunoregulatory proteins (mannose receptor C-type 1 [Mrc1] and ceramide synthase 2 [CerS2]) and importantly contributed to the creation of an immunosuppressive microenvironment in inflamed and fibrotic lungs46,50.
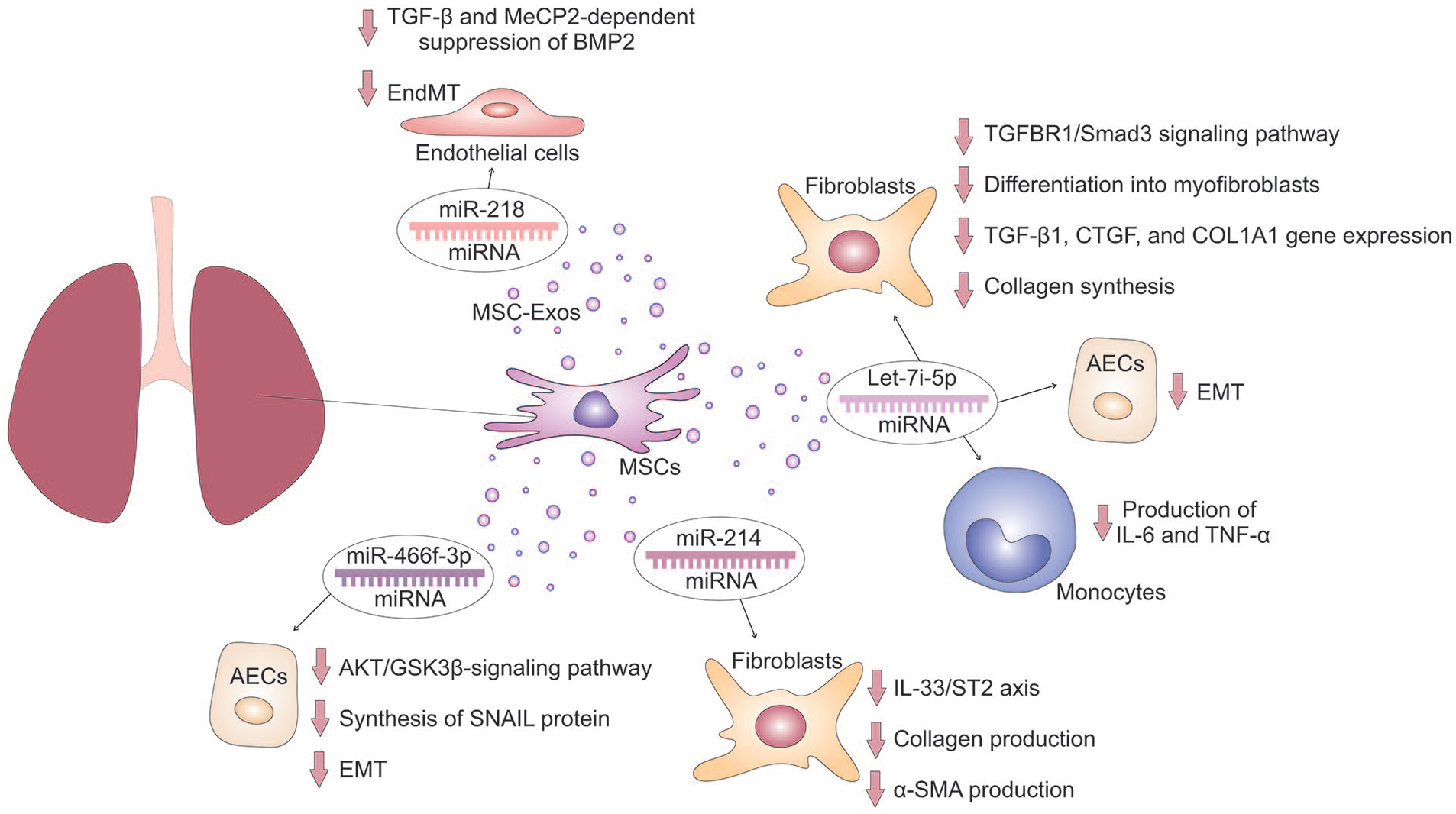
MSC-Exos are also enriched with MSC-miRNAs, which modulate protein synthesis in target AECs, myofibroblasts, and lung-infiltrated immune cells through the post-transcriptional regulation of target mRNA 16 . MSC-miRNAs are MSC-derived, small, non-coding RNA molecules which consist of 20–22 base sequences 17 . They have seed regions (nucleotide sites 2–8) that bind to target mRNA, leading to its degradation or inhibiting its translation [Fig. 1]16,17. This binding to mitochondria-related mRNA impairs the production of proteins in target cells: inflammatory and pro-fibrotic cytokines in immune cells, pro-apoptotic molecules in injured AECs, and pro-fibrotic transcriptional factors in activated fibroblasts, preventing their differentiation in myofibroblasts17,18. MSC-miRNAs can also directly bind to target proteins, altering their function, and bind to other non-coding RNAs, negatively regulating their functions 17 . In lung-infiltrated DCs and alveolar macrophages, MSC-miRNAs can suppress TLR–dependent signaling, resulting in attenuated activation of NLRP3 inflammasome and in decreased production of NLRP3-dependent inflammatory cytokines (IL-1β and IL-18) 16 . By affecting synthesis of cyclin-dependent kinases that regulate cell cycle, MSC-miRNAs may improve viability and survival of injured AECs and may prevent the expansion of activated, inflammatory Th1, Th1, and Th17 lymphocytes, suppressing the progression of Th1/Th2/Th17 cell-driven lung inflammation16,17. Therefore, results in several recently published experimental studies provided evidence about beneficial effects of MSC-derived Let-7i-5p, miR-218, miR-214, and miR-466f-3p in the treatment of pulmonary fibrosis (Fig. 2)51–54.
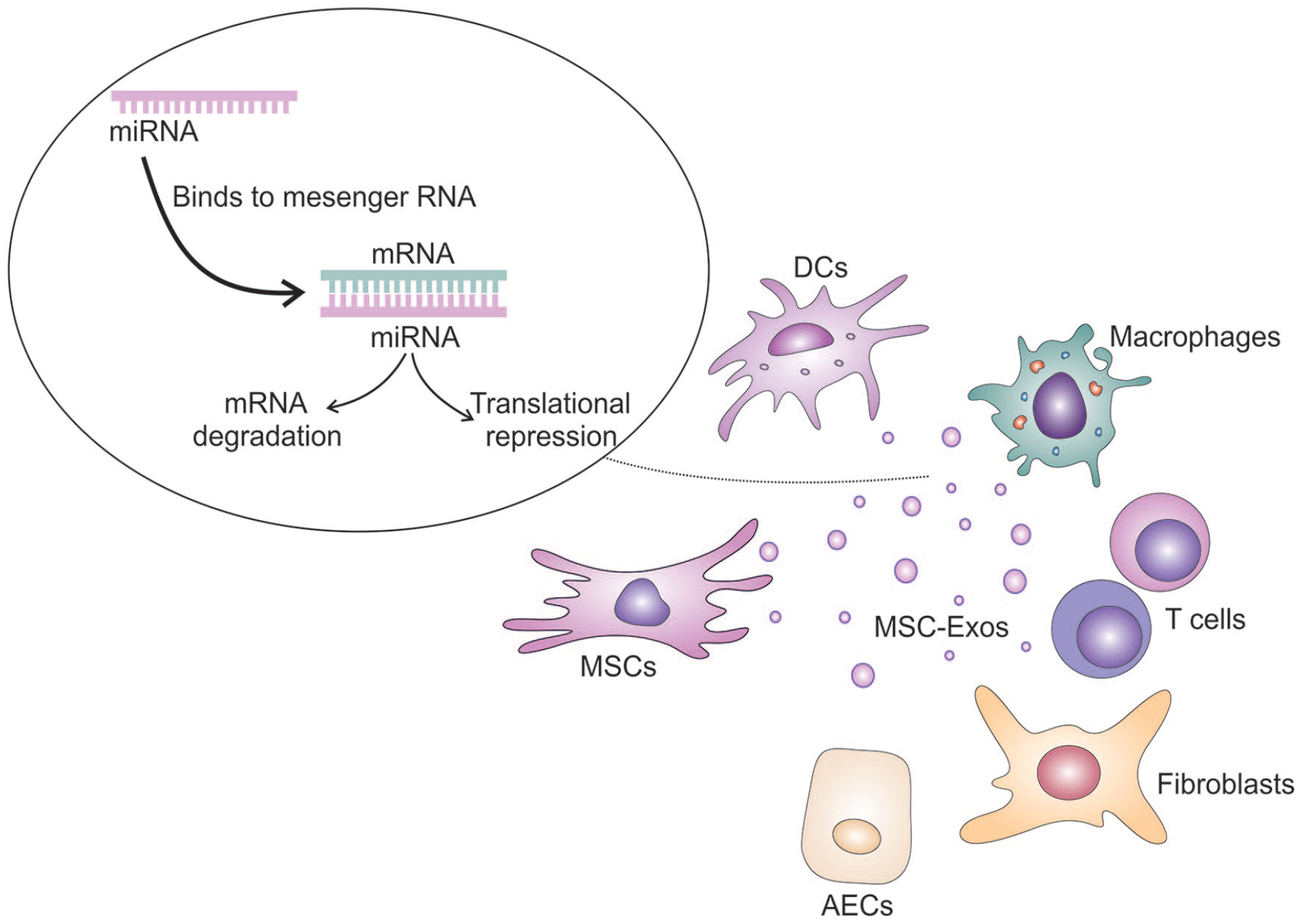
MSC-miRNAs–dependent regulation of protein production. MSC-miRNAs are contained in MSC-Exos. Within MSC-Exos, MSC-miRNAs are delivered in target immune cells and AECs. MSC-miRNAs modulate protein synthesis in AECs, fibroblasts, and lung-infiltrated immune cells through the post-transcriptional regulation of target mRNA. Also, MSC-miRNAs have seed regions that bind to target mRNA, leading to its degradation or inhibiting its translation.
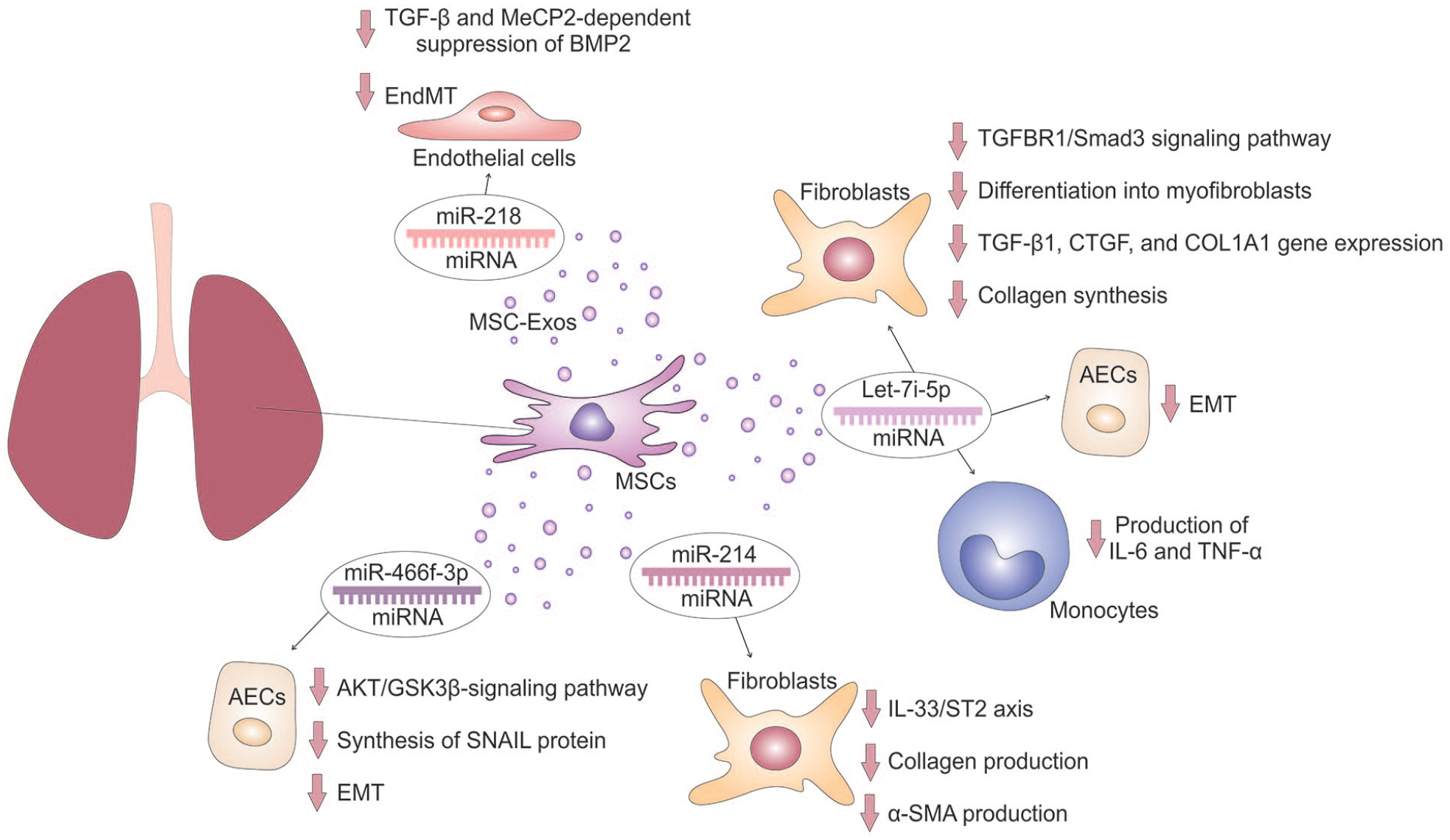
MSC-miRNAs–dependent attenuation of pulmonary fibrosis. MSC-derived Let-7i-5p collagen synthesis suppresses TGFBR1/Smad3 signaling pathway and down-regulates TGF-β1, CTGF, and COL1A1 gene expression in fibroblasts, preventing their differentiation into myofibroblasts. In addition, MSC-miRNAs attenuate production of IL-6 and TNF-α in monocytes and modulates the phenotype and function of AECs, preventing EMT. MSC-sourced miR-218 inhibits TGF-β and MeCP2-dependent suppression of BMP2 in endothelial cells and prevents EndMT. MSC-derived miR-214 suppresses the IL-33/ST2 axis in fibroblasts, attenuating collagen and α-SMA production. MSC-sourced miR-466f-3p down-regulates AKT/GSK3β-signaling pathway and attenuates the synthesis of SNAIL protein in AECs, preventing EMT and lung fibrosis.
Molecular Mechanisms Responsible for MSC-miRNAs–Dependent Attenuation of Lung Fibrosis
MSC-sourced Let-7i-5p is a member of the Let-7 family of miRNA, which are well known for their regulatory functions in various biological processes, such as cell proliferation, differentiation, and apoptosis 51 . The “5p” in Let-7i-5p indicates that this miRNA is derived from the 5' end of the precursor molecule and is typically the guide strand involved in target gene regulation. MSC-derived Let-7i-5p has been found to have diverse target genes in different cell types, and its deregulation has been associated with various inflammatory, fibrotic, and malignant diseases 51 . Xu and colleagues 51 used an animal model of silica-induced pulmonary fibrosis to investigate the therapeutic potential of hUC-MSC–derived Let-7i-5p in the attenuation of lung fibrosis. Obtained results showed that hUC-MSC–sourced Let-7i-5p inhibited the TGFBR1/Smad3 signaling pathway in lung fibroblasts, thereby preventing their activation and subsequent differentiation into myofibroblasts. This mechanism involved the suppression of TGF-β1, CTGF, and COL1A1 gene expression, which reduced excessive collagen production and extracellular matrix deposition in the inflamed lungs 51 . In addition, hUC-MSC–derived Let-7i-5p induced apoptosis and senescence in fibroblasts and myofibroblasts, limiting their ability to contribute to fibrosis 51 . Moreover, Let-7i-5p inhibited EMT by targeting Snail and Twist proteins, thereby preserving the integrity of the epithelial barrier and preventing leukocyte infiltration into the lungs 51 . In inflammatory monocytes infiltrating the lungs, MSC-derived Let-7i-5p suppressed the production of pro-inflammatory cytokines (IL-6 and TNF-α), thus preventing inflammation-driven tissue damage and fibrosis 51 .
MSC-derived miR-218 is a specific miRNA that has been shown to play various roles in cellular processes such as development, differentiation, and tissue homeostasis54–56. It has been found to inhibit the proliferation and migration of cancer cells, making it a potential target for cancer treatment 56 . In addition, MSC-derived miR-218 has been implicated in neurogenesis and neuroprotective effects, suggesting its potential for neurological disorders 56 . Having in mind its potential for the regulation of cell differentiation, Zhao and colleagues 52 investigated molecular mechanisms responsible for MSC-sourced miR-218-dependent modulation of endothelial-to-mesenchymal transition (EndMT), which plays important pathogenic role in the development of pulmonary fibrosis. During EndMT, ECs undergo a transformation into mesenchymal cells, acquiring migratory and invasive properties 57 . This process involves the loss of endothelial characteristics, upregulation of mesenchymal markers (α-SMA and fibroblast specific protein 1 (FSP1)), migration into the surrounding tissue, and promotion of ECM protein production and fibroblast activation 57 . Signaling pathways such as TGF-β, BMP2, and MeCP2 are involved in driving EndMT and pulmonary fibrosis57,58. In bleomycin-induced pulmonary fibrosis, continuous activation of TGF-β, and repression of BMP2-driven signaling worsen the fibrotic response52,57,58. MeCP2, an activator of α-SMA expression, contributes to lung fibrosis by suppressing the BMP2 pathway57,58. Zhao and colleagues 52 demonstrated that hUC-MSC–derived miR-218 attenuated bleomycin-induced pulmonary fibrosis by preventing EndMT. Zhao et al. 52 found that hUC-MSC–sourced miR-218, contained within MSC-Exos, restored endothelial properties weakened by TGF-β and MeCP2-dependent suppression of BMP2. Knockdown of miR-218 in MSCs partially reversed the beneficial effects of MSC-Exos, while transfection of miR-218 and its overexpression in MSC-Exos increased BMP2 expression, leading to the attenuation of EndMT and alleviation of pulmonary fibrosis 52 .
MSC-derived miR-214 is a versatile miRNA with a wide range of functions in different biological processes, including cardiovascular diseases, immune responses, and neural development 59 . It is involved in regulating smooth muscle cell proliferation, migration, and differentiation 59 . Aberrant expression of miR-214 has been observed in atherosclerosis and cardiac hypertrophy 60 . It can modulate the expression of genes involved in cell cycle regulation, collagen synthesis, and extracellular matrix remodeling59,60. In addition, MSC-derived miR-214 is involved in regulating immune responses 61 . It can modulate the differentiation and function of various immune cell types, including T cells, macrophages, and DCs 61 . By targeting immune-related genes, miR-214 can influence immune cell activation, cytokine production, and antigen presentation 61 . In line with these findings, Xie and colleagues 53 recently discovered that human BM-MSC–derived miR-214 can inhibit the progression of bleomycin-induced pulmonary fibrosis. This is achieved by suppressing the IL-33/ST2 axis in the fibrotic lungs 53 . IL-33 is an alarmin released from injured cells in the lungs during an inflammatory response 62 . It promotes lung fibrosis by recruiting and activating inflammatory immune cells, inducing the secretion of pro-fibrotic cytokines, activating fibroblasts, and promoting EMT 62 . Xie and colleagues 53 found that MSC-derived miR-214 targets IL-33 and blocks its interaction with ST2 receptor in fibroblasts, leading to a reduction in collagen fiber accumulation and α-SMA production, ultimately attenuating pulmonary fibrosis in mice.
Similar findings were reported by Li and colleagues, 54 who demonstrated that mouse BM-MSC–sourced miR-466f-3p can attenuate radiation-induced lung fibrosis by suppressing EMT. MSC-derived miR-466f-3p is expressed in various tissues during embryonic development 63 . It has been shown to promote the differentiation of cells into specific cell types by targeting specific mRNA molecules involved in maintaining the stem cell state 63 . In addition, MSC-sourced miR-466f-3p regulates the immune response by targeting mRNA molecules involved in activation and differentiation of T cells 64 . It also modulates the expression of mRNAs that are responsible for synthesis of co-stimulatory molecules and inflammatory cytokines in macrophages and DCs, thereby influencing the overall immune response 65 . Li and colleagues 54 demonstrated that mouse BM-MSC–derived miR-466f-3p inhibited collagen production in the lungs, crucially contributed to the attenuation of pulmonary fibrosis. Mechanistically, it inhibits the synthesis of a protein called SNAIL by suppressing the protein kinase B (PKB)/Glycogen synthase kinase 3 beta (GSK3β)-signaling pathway in a c-MET-dependent manner 54 . SNAIL is a transcription factor involved in EMT development in the lungs 66 . Activation of SNAIL leads to various pathological changes, including disruption of the epithelial barrier, infiltration of inflammatory cells and fibrogenic mediators, activation of fibroblasts, excessive extracellular matrix deposition, and enhanced fibroblast migration and contractility 66 . SNAIL is up-regulated in response to pro-fibrotic TGF-β, and its activation is regulated by PKB and GSK3β kinases5,66. Inhibition of PKB releases the repression of GSK-3β, leading to down-regulation of SNAIL in inflamed and fibrotic lungs 66 . By modulating the PKB/GSK3β-signaling pathway, MSC-derived miR-466f-3p suppressed SNAIL activity, prevented EMT, and attenuated pulmonary fibrosis in irradiated mice 54 .
Conclusions and Future Perspectives
MSC-derived anti-fibrotic and immunoregulatory miRNAs are delivered directly in target immune cells, AECs, and fibroblasts by MSC-EVs16,17. Biocompatibility, low immunogenicity, and long-circulating half-life make MSC-EVs promising candidates for various biomedical applications and regenerative medicine approaches42,67. As they originate from MSCs, MSC-EVs are biocompatible and well-tolerated 42 . In addition, MSC-EVs exhibit low immunogenicity because they lack MHC class II molecules, reducing the likelihood of triggering allogeneic immune response in MHC class II mismatched recipients 67 . Importantly, MSC-EVs have a long-circulating half-life, allowing them to remain in the bloodstream for an extended period, facilitating their therapeutic effects and potential for targeted delivery to specific tissues or organs 67 .
Nevertheless, it should be noted that many challenges should be addressed prior to widespread clinical application of MSC-derived EVs 68 . Most importantly, EVs are known to exhibit variability based on the technique used for their isolation 68 . For example, ultracentrifugation may yield a different EV population compared to size exclusion chromatography or polymer-based precipitation methods 68 . Each isolation technique can selectively enrich specific subtypes of EVs based on their size, density, or surface markers. In order to address this issue, specific standardization guidelines for isolation and quantification of MSC-EVs have to be established and followed in order to align various aspects of MSC-EV biology and therapeutic effectiveness with their quantifiable characteristics 68 . For example, MSC-EVs should be described using measurable criteria to determine their cellular origin, the presence of lipid-membrane vesicles, and the level of structural and biochemical integrity. All newly isolated MSC-EVs must be compared against a well-documented MSC-EVs biological standard prior to their clinical use 68 .
Among various MSC-Exo-derived miRNAs that may affect proliferation, activation, and production of inflammatory and pro-fibrotic cytokines in lung-infiltrated immune cells, only MSC-sourced Let-7i-5p, miR-214, miR-218, and miR-466f-3p showed beneficial effects in the attenuation of pulmonary fibrosis in experimental settings51–54. These miRNAs could also prevent EMT, EndMT, promote survival of injured AECs, inhibit activation and differentiation of fibroblast, and suppress production of collagen in myofibroblasts51–54.
It should be noted that, in addition to their anti-inflammatory and anti-fibrotic effects, MSC-derived Let-7i-5p and miR-218 may act as tumor suppressors69,70. Let-7i-5p is a negative regulator of cell proliferation since it specifically targets transcriptional factors involved in cell growth and division. By inhibiting the expression of growth-promoting genes, Let-7i-5p helps maintain normal cell growth and prevents the formation of tumors 69 . Similarly, by down-regulating synthesis of pro-angiogenic proteins in tumor ECs, miR-218 suppresses EC sprouting, tube formation, and neo-angiogenesis in tumors, attenuating their growth and metastasis 70 .
However, opposite to Let-7i-5p and miR-218, other anti-fibrotic MSC-sourced miRNAs (miR-214 and miR-466f-3p) may either promote or suppress tumor growth, depending on the specific cancer type and cellular context71,72. Results obtained in animal models of breast and colon cancer showed that MSC-sourced miR-214 and miR-466f-3p may act as tumor suppressors by targeting mRNA molecules that promote growth, invasion, and metastasis of malignant cells71,72. Conversely, these miRNAs can promote tumor growth and progression by down-regulating tumor-suppressor genes or by affecting the expression of genes involved in apoptosis of cancer cells71,72. In line with these findings, up-coming experimental studies have to determine “pro- or anti-tumorigenic” potential for MSC-derived Let-7i-5p, miR-214, miR-218, and miR-466f-3p in the injured, inflamed, and fibrotic lungs, before these anti-fibrotic molecules could be offered as new remedies in the treatment of pulmonary fibrosis.
Footnotes
Authors’ Contributions
Conceptualization: C.R.H. and V.V.; Writing—original draft preparation: A.V., A.A., and V.V.; Writing—review and editing: C.R.H. and V.D.; Design and preparation of figures: A.A.; Funding acquisition: V.V. All authors have read and agreed to the final version of the manuscript.
Availability of Data and Material
The data that are discussed in this article are presented in cited studies.
Ethical Approval
This study was approved by our institutional review board.
Statement of Human and Animal Rights
This article does not contain any studies with human or animal subjects.
Statement of Informed Consent
There are no human subjects in this article, and informed consent is not applicable.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by Serbian Ministry of Science (grant no. 451-03-47/2023-01/200111) and Faculty of Medical Sciences University of Kragujevac (grant no. MP01/18).