
Abstract
Human induced pluripotent stem cells (iPSCs) have already been used in transplantation therapies. Currently, cells from healthy people are transplanted into patients with diseases. With the rapid evolution of genome editing technology, genetic modification could be applied to enhance the therapeutic effects of iPSCs, such as the introduction of secreted molecules to make the cells a drug delivery system. Here, we addressed this possibility by utilizing a Fabry disease mouse model, as a proof of concept. Fabry disease is caused by the lack of α-galactosidase A (GLA). We previously developed an immunotolerant therapeutic molecule, modified α-N-acetylgalactosaminidase (mNAGA). We confirmed that secreted mNAGA from genome-edited iPSCs compensated for the GLA activity in GLA-deficient cells using an in vitro co-culture system. Moreover, iPSCs transplanted into Fabry model mice secreted mNAGA and supplied GLA activity to the liver. This study demonstrates the great potential of genome-edited iPSCs secreting therapeutic molecules.
Introduction
Induced pluripotent stem cells (iPSCs) have great potential as resources for cell therapy 1 . Since the first clinical trial of iPSC-based transplantation into an age-related macular degeneration patient in 2014 2 , several transplantation therapies using iPSC-derived platelets (jRCTa050190117), corneal epithelial cell sheets (jRCTa050190084), cardiomyocyte sheets (jRCT2053190081), and dopaminergic progenitors (UMIN000033564) have proceeded to clinical trials.
So far, these transplantations have utilized unmodified iPSCs from healthy subjects 3 . However, we can potentially improve the therapeutic effects of iPSCs by genome editing as long as the safety is guaranteed4–6. For example, disruption of some of the human leukocyte antigen (HLA) genes can expand the range of people who can receive the cells without immune responses 3 .Genomic insertion of chimeric antigen receptor has been utilized for T-cell immunotherapy 7 . Another prospect of genetic modification is to make iPSCs that secrete therapeutic molecules. Recently, the implantation of genome-engineered iPSCs that secrete interleukin-1 in rheumatoid arthritis mice demonstrated the potential of this strategy 8 . However, the transplantation of iPSCs to deliver therapeutic molecules in vivo has not been established as a therapeutic option. Therefore, we decided to test whether iPSCs with genetic modifications can deliver therapeutic molecules in vivo in mice by focusing on Fabry disease as a proof of concept.
Fabry disease (OMIM 301500) is an X-linked genetic disorder 9 that is caused by a deficiency of α-galactosidase A (GLA, EC 3. 2. 1. 22). GLA is a lysosomal enzyme that is partially secreted, and incorporated by other cells. Therefore, the loss of GLA results in accumulation of its substrates such as globotriaosylceramide (Gb3) and globotriaosylsphingosine (Lyso-Gb3) 10 . The accumulation leads to various symptoms, including cardiomyopathy, renal failure, and stroke 11 .
Currently, there are two major treatments for Fabry disease: enzyme replacement therapy (ERT)12,13 and pharmacological chaperone therapy (PCT) 14 . In ERT, the recombinant human GLA is intravenously infused every 2 weeks. However, this infusion can lead to the development of antibodies against GLA and hampers the therapeutic effect in some patients 15 . In PCT, small molecules that stabilize GLA are orally administered. However, PCT has no effect in patients with the complete loss of GLA16,17.
To solve these problems, we aimed to establish a new iPSC transplantation therapy for Fabry disease using a modified enzyme that we previously developed, modified α-N-acetylgalactosaminidase (mNAGA). mNAGA was created by altering the substrate specificity of NAGA, which is a paralog of GLA, into that of GLA 18 . Because mNAGA maintains the original antigenicity, this modified enzyme has no immunological cross-reactivity with GLA, while having the GLA enzymatic activity. In this study, we tested if transplantation of iPSCs secreting mNAGA by genome editing could supply the GLA activity in vivo. Our strategy of cell therapy using genome-edited iPSCs can be applied to various diseases caused by loss of functional molecules.
Materials and Methods
Genome Editing of Human iPSCs
In this study, an iPSC line from a healthy subject, WTC11 (GM25256 from Coriell Institute), was used 19 . For disruption of GLA, we used the P3 Primary Cell Kit and Nucleofector 4D device (Lonza, Basel, Switzerland). Two hundred thousand WTC11 iPSCs were transfected with 500 ng of pX330 with a guide RNA (gRNA) targeting the first exon of the GLA gene (5′-GTTCCTCAGCTGCATTGTCA-3′) by the DS-138 program. Induced indels were detected by Droplet Digital PCR, and clones with indels were isolated by sib-selection (repeated limited dilutions), as described previously 20 .
For knock-in of mNAGA complementary DNA (cDNA) driven by the CAG promoter, we used the Human Stem Cell Nucleofector Kit-1 and Nucleofector 2b Device (Lonza). Two million iPSCs were transfected with 2 µg of each AAVS1-TALEN-L/R (Addgene #59025 and #59026) 21 and 5 µg of AAV-CAGGS-mNAGA donor plasmid (Addgene #196448). AAV-CAGGS-mNAGA was constructed by replacing the EGFP cDNA of AAV-CAGGS-EGFP (Addgene #22212) 22 with mNAGA cDNA. After transfection using Nucleofector 2b using the A-23 program, the treated cells were resuspended in mTeSR Plus with Y-27632 and plated into a Matrigel-coated 96-well plate. Two days after transfection, the medium was exchanged with mTeSR Plus with Y-27632 and 0.5 µg/ml Puromycin for selection of transfected cells. The cells were cloned by limiting dilution. We extracted genomic DNA from cloned cells and confirmed the knock-in by junction polymerase chain reaction (PCR) and then Sanger sequencing.
Measurement of the GLA Activity Using an Artificial Fluorogenic Substrate
Hydrolysis of an artificial GLA substrate, 4-methylumbelliferyl-α-
The Mass Spectrometric Analysis of Gb3 and Lyso-Gb3
For measurement of Gb3 and Lys-Gb3 in organs and tissues of mice, a liquid chromatography (LC)-tandem mass spectrometry (MS/MS) analysis was performed according to a previous report 24 . Briefly, the organs and tissues were homogenized in MES buffer, and a 10-µl aliquot of the homogenate was mixed with a 70-µl aliquot of chloroform: methanol (1:2). Then a 10-µl aliquot of 5 µg/ml Gb3 (C17:0; Matreya, LLC, State College, Pennsylvania, United States) and a 10-µl aliquot of 500 nmol/l stable isotope-labeled (one 13C and three deuteriums) Lyso-Gb3 (Nard Institute, Kobe, Japan) were added as internal standards. The mixture was centrifuged and the supernatant was transferred to an LC vial. For LC, a Union UK-C8 column (20 × 3 mm ID., 3 µm; Imtakt Co., Portland, United States) was used, and the column oven was set at 30°C. Chromatographic separation was performed with a binary gradient consisting of a mobile phase of water containing 0.1% acetic acid and 2 mmol/l ammonium acetate and methanol containing 0.1% acetic acid and 2 mmol/l ammonium acetate. The flow rate was 0.25 ml/min and the injection volume was 2 μl. Then, Gb3 isoforms and Lyso-Gb3 in the samples were detected by MS/MS using a LCMS-8040 triple quadrupole mass spectrometer (Shimadzu, Kyoto, Japan) equipped with an electrospray ionization interface in positive-ion mode. The multiple reaction monitoring (MRM) conditions were optimized with an automatic MRM optimization function. The calculations for measurement of Gb3 isoforms and Lyso-Gb3 were performed using LabSolutions (Shimadzu), and the Gb3 contents of organs and tissues were calculated from the sums of those of the Gb3 isoforms.
Co-culture of Two Types of Cells Using Trasnswells
We used the bottom dish and the insert for GLA-deficient cells and mNAGA-supplier cells, respectively (Fig. 2). These cells were plated in the bottom dish or the insert and separately cultured until the cells reached 100% confluence. Then, the insert was moved to the bottom dish for co-culture. For undifferentiated iPSCs, both the bottom dish and the insert were coated with Matrigel. We co-cultured for 3 days for mouse fibroblasts, and for 1 week for other cell types. We chose the medium for co-culture depending on the GLA-deficient cell types in the bottom dish.
Transplantation of Human iPSCs into Mouse Testes
iPSCs were detached using 500 µl of 0.5 mM ethylenediaminetetraacetic acid (EDTA) (Nacalai Tesque, Kyoto, Japan) in phosphate buffered saline (PBS) per well of a 6-well plate and resuspended in 1.5 ml of mTeSR Plus with 10 µM Y-27632. We used a Cell Lifter (Greiner) to scrape the cells while keeping the cell clumps. One million cells were collected into a 1.5 ml microtube and centrifuged at 200 × g for 3 min at room temperature. We removed the supernatant and tapped the cell pellets. A mixture of 50 µl of growth factor reduced (GFR) Matrigel Phenol Red-Free and 50 µl of mTeSR Plus with 10 µM Y-27632 was added and gently mixed with the cells. The cell suspension was aspirated by a 1-ml syringe (Terumo, Shibuya, Japan) with an 18G needle (Terumo) and stored on ice until transplantation. Mice were anesthetized by 2% isoflurane (Viatris, Canonsburg, Pennsylvania, United States). A 1-cm incision was made on the abdominal region, and the epididymal fat pad was carefully pulled out along with the testis. The cell suspension was injected using a syringe and held for 10 s to avoid backflow. The testis was pushed back to the origin location, and the incision was sutured.
Statistical Analyses
Data are shown as the mean ± standard error of the mean in all graphs generated using the Microsoft Excel software program (version 16.66 for Mac; Microsoft, Redmond, Washington, United States). For comparison of two samples, P-values were determined by an unpaired two-tailed Student’s t-test using the Microsoft Excel software program (version 16.66 for Mac; Microsoft).
Results
Generation of Human iPSCs Secreting mNAGA by Genome Editing
First, we established a human iPSC line that secretes mNAGA to test its therapeutic effects in Fabry model mice. To exclude the possible immunogenic reactions caused by the endogenous GLA of iPSCs in patients, we knocked down GLA by introducing a 6-bp deletion including the start codon by CRISPR-Cas9 (Fig. 1A; hereafter called GLA-KO iPSCs). We confirmed the loss of GLA activity in GLA-KO iPSCs (Fig. 1B). We also sequenced 22 most likely off-target sites predicted by Cas-OFFinder 25 in GLA-KO iPSCs and found no off-target effects (Supplemental Fig. S1).

Establishment of mNAGA-secreting iPSCs. (A) Design of the disruption of the GLA gene in human iPSCs by CRISPR-Cas9. We introduced a 6-bp deletion containing the start codon. The cut site and start codon are shown by red triangles and red letters, respectively. Sanger sequencing data around the deletion are shown. (B) GLA activity in the lysates or culture supernatants of WT, GLA-KO, and GLA-KO+mNAGA iPSCs (n = 3). **P < 0.01. (C) Design of knock-in of mNAGA cDNA driven by the CAG promoter into AAVS1 using TALENs. cDNA: complementary DNA; TALENs: transcription activator-like effector nucleases; PAM: protospacer adjacent motif; NHEJ: non-homologous end joining; gRNA: guide RNA; SA: splice acceptor; mNAGA: modified α-N-acetylgalactosaminidase; iPSCs: induced pluripotent stem cells; GLA: galactosidase A; AAVS1: adeno-associated virus integration site 1; WT: wild type.
Next, to establish an iPSC line stably expressing mNAGA, we heterozygously knocked in mNAGA cDNA driven by the CAG promoter into adeno-associated virus integration site 1 (AAVS1), a safe harbor locus, by transcription activator-like effector nucleases (TALENs) previously reported to have no detectable off-target effects 26 (Fig. 1C; hereafter called GLA-KO+mNAGA iPSCs). This cell line exhibited higher GLA activity than wild type (WT) and GLA-KO iPSCs both in cell lysate and in culture supernatant (Fig. 1B). Thus, we successfully generated iPSCs for a potential cell therapy for Fabry disease by genome editing.
mNAGA Secreted from iPSCs Was Taken Up by GLA-Deficient Cells in vitro
For cell therapy, mNAGA secreted from transplanted iPSCs must be taken up by patients’ cells. Therefore, we investigated whether GLA-deficient cells could pick up mNAGA secreted from iPSCs by co-culture using transwells (Fig. 2A).
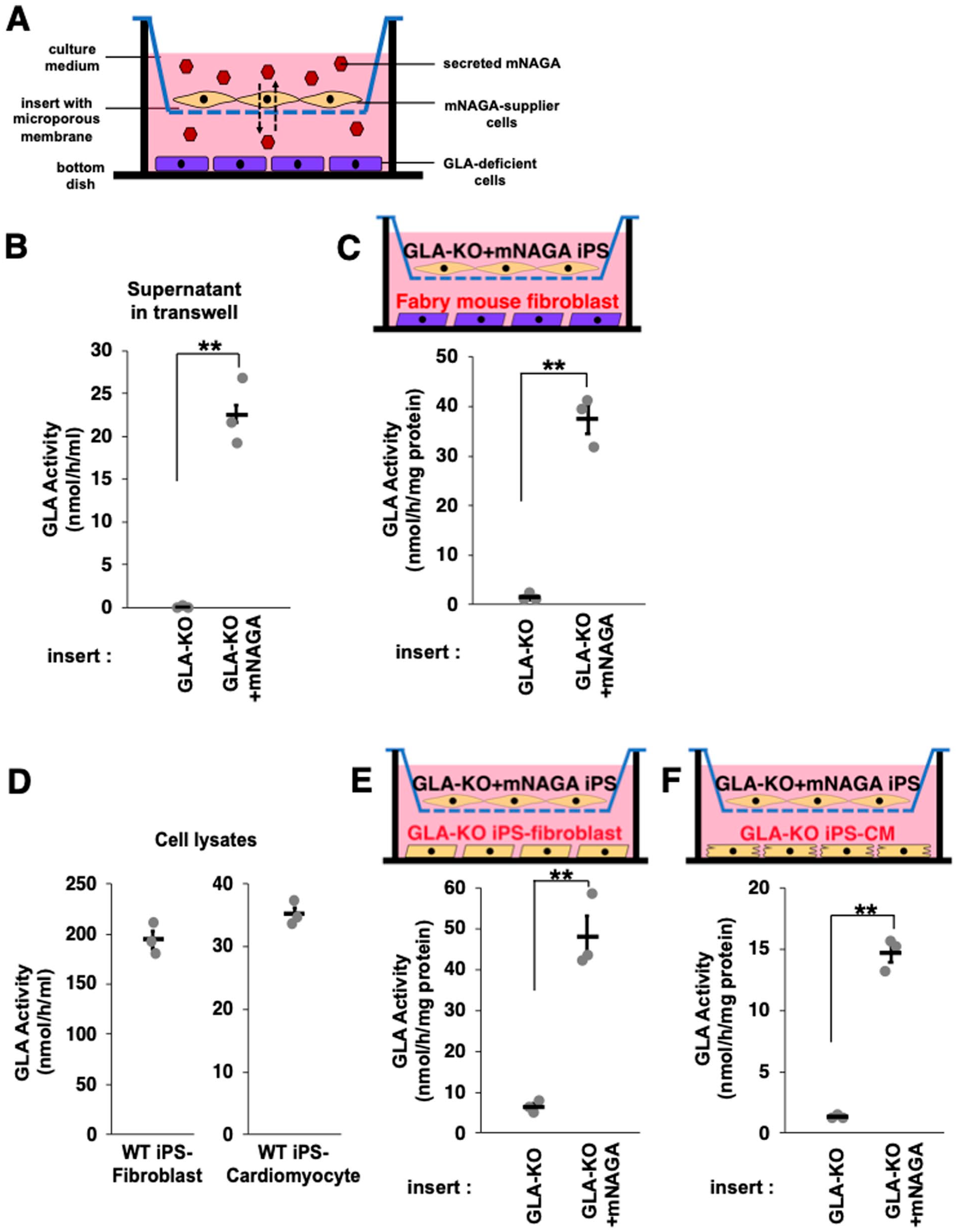
In vitro delivery of mNAGA secreted from iPSCs to GLA-deficient cells. (A) Schematic illustration of in vitro delivery of mNAGA using transwells. (B) GLA activity of the culture supernatants of GLA-KO or GLA-KO+mNAGA iPSCs (n = 3). **P < 0.01 (C) GLA activity of mouse fibroblast lysates co-cultured with GLA-KO or GLA-KO+mNAGA iPSCs (n = 3). *P < 0.05 and **P < 0.01. (D) GLA activity of WT iPSC–derived fibroblast and CM (n = 3). (E, F) GLA activity of GLA-KO iPSC–derived fibroblasts (E) and GLA-KO iPSC–derived CMs (F) co-cultured with GLA-KO or GLA-KO+mNAGA iPSCs (n = 3). *P < 0.05 and **P < 0.01. mNAGA: modified α-N-acetylgalactosaminidase; iPSCs: induced pluripotent stem cells; GLA: galactosidase A; WT: wild type; CM: cardiomyocyte.
First, we investigated whether mNAGA secreted from iPSCs can be incorporated into fibroblasts derived from Fabry mice without functional GLA, as described previously 27 . We co-cultured Fabry mouse fibroblasts on the bottom of a transwell and undifferentiated GLA-KO or GLA-KO+mNAGA iPSCs on the insert for 3 days. We confirmed the secretion of mNAGA from the iPSCs by measuring the GLA activity in the culture medium (Fig. 2B). As expected, the GLA activity in the co-cultured Fabry mouse fibroblasts was higher than in the control. These findings indicated that secreted mNAGA restored the GLA activity of Fabry mouse fibroblasts (Fig. 2C).
Next, to simulate situations more relevant to actual cell therapy, we co-cultured GLA-KO iPSC–derived fibroblasts or cardiomyocytes with GLA-KO+mNAGA iPSCs. Compared with the GLA activity of WT cells (Fig. 2D), we observed the restoration levels of the GLA activity in GLA-KO iPSC–derived fibroblasts and cardiomyocytes by co-culturing were 24.7% and 41.8%, respectively (Fig. 2E, F). These results demonstrated that mNAGA secreted from GLA-KO+mNAGA iPSCs can be incorporated into mouse and human GLA–deficient cells and complement the lost GLA activity.
Generation of an Immunodeficient Fabry Mouse Model
Next, we generated an immunodeficient Fabry mouse model in which human cells can be transplanted and engrafted. We disrupted the Gla gene in NOD.CB17-Prkdcscid/J (NOD SCID) mice, a mouse strain without functional T and B cells 28 by injecting Cas9 protein and single guide RNA (sgRNA) targeting exon1 of mouse Gla (Fig. 3A). We obtained 25 founder mice with various insertions and/or deletions out of 227 injected and transferred zygotes (Table 1). Among them, we established one mouse line with a 28-bp deletion (GlaΔ28) spanning the start codon of Gla (Fig. 3A). We confirmed that the GLA activity was lost in the liver, heart, kidney, and blood plasma of this GlaΔ28 mouse strain (Fig. 3B).

Generation of the immunodeficient Fabry mouse model. (A) Design of the disruption of Gla in NOD SCID mouse zygotes. The cut site and the start codon are shown by red triangles and red letters, respectively. The allele with a 28-bp deletion containing the start codon identified as a null allele, GlaΔ28, is shown. The Sanger sequencing data show the WT allele and the 28-bp deletion allele in a heterozygous mouse. (B) The GLA activity in the liver, heart, kidney, and blood plasma of male WT and GlaΔ28 mice (n = 6). **P < 0.01. (C) Accumulation of Gb3 and Lyso-Gb3, substrates of GLA, in the liver, heart, and kidneys of male WT and GlaΔ28 mice (n = 6). **P < 0.01. PAM: protospacer adjacent motif; NHEJ: non-homologous end joining; gRNA: guide RNA; GLA: galactosidase A; WT: wild type; Gb3: globotriaosylceramide; Lyso-Gb3: globotriaosylsphingosine.
Edited Gla Alleles of NOD SCID Newborn Mice.

All sequences start from 5' side. Only the sequence of male7 with 98-bp deletion starts from 91-bp upstream of the open reading frame. Open reading frames are highlighted in capital letters. Deletions and insertions are shown in null bars and red letters, respectively. WT: wild type.
Next, we measured the amounts of Gb3 and Lyso-Gb3, substrates of GLA, in the GlaΔ28 mouse strain by MS/MS. As we expected, these substrates were accumulated in all analyzed tissues (Fig. 3C). These results confirmed that the NOD SCID GlaΔ28 mouse strain we established could serve as an immunodeficient Fabry mouse model.
Transplantation of GLA-KO+mNAGA iPSCs to NOD SCID Fabry Mice
Finally, we transplanted GLA-KO+mNAGA iPSCs into the NOD SCID Fabry mice. As the most efficient strategy to transplant the largest number of human cells, we formed teratomas in these mice.
We injected 1.0 × 106 undifferentiated GLA-KO or GLA-KO+mNAGA iPSCs into the testes of 7- or 8-week-old male NOD SCID GlaΔ28 mice. After 7 or 8 weeks, we observed teratomas in all treated mice (Fig. 4A and Supplemental Fig. S2). We confirmed that GLA-KO+mNAGA iPSC–derived teratoma maintained the GLA activity (Fig. 4B).
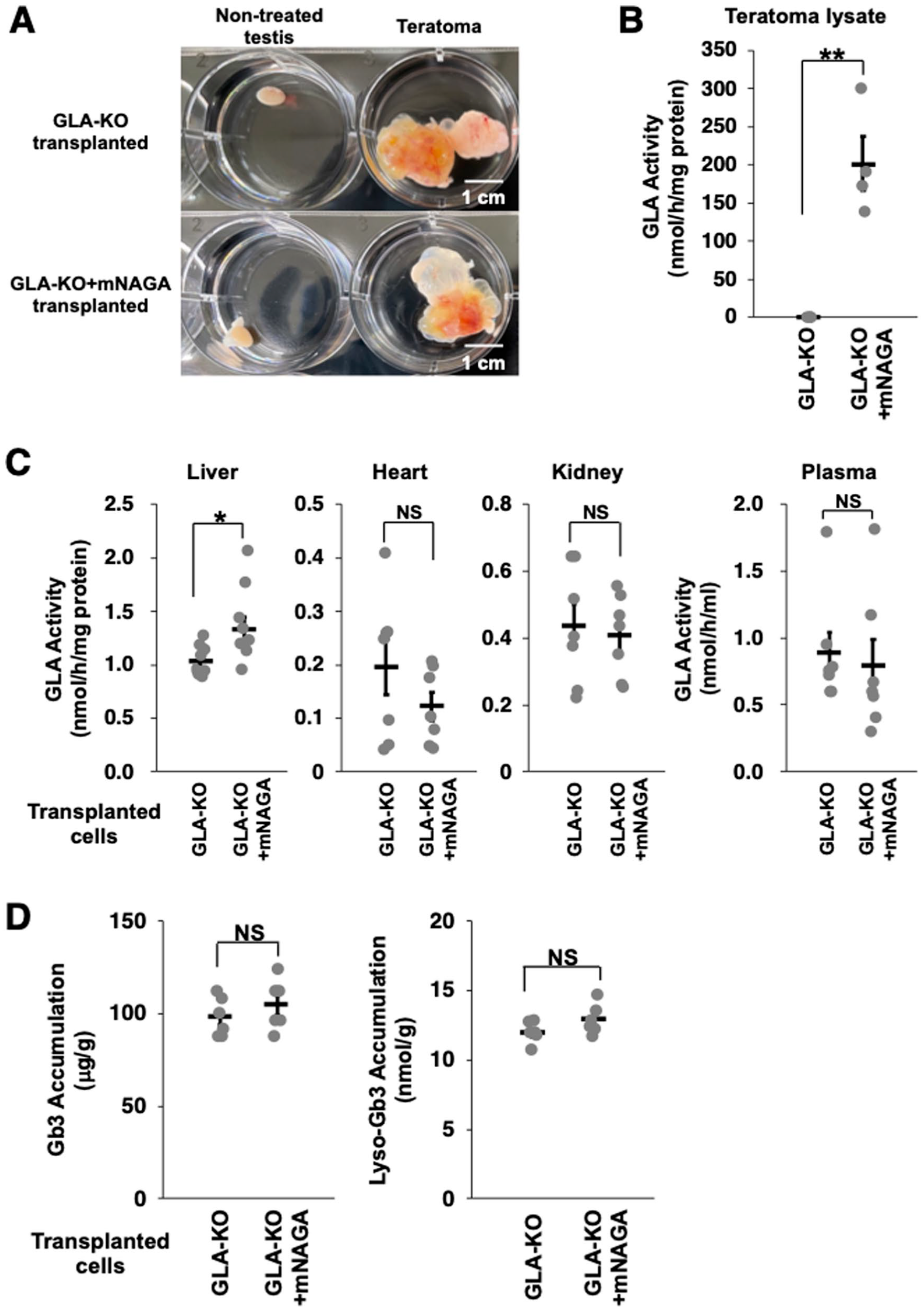
Transplantation of GLA-KO+mNAGA iPSCs to NOD SCID Fabry model mice. (A) Images of a non-treated testis and a testis with teratoma. Scale bars indicate 1 cm. (B) GLA activity in teratoma lysates derived from GLA-KO or GLA-KO+mNAGA iPSCs (n = 4). *P < 0.05 and **P < 0.01. (C) GLA activity in the liver, heart, kidney, and blood plasma of NOD SCID Fabry mice transplanted with GLA-KO or GLA-KO+mNAGA iPSCs (n = 9 in only liver; n = 6 in heart, kidney, and blood plasma). P < 0.05, **P < 0.01, and NS (P > 0.1). (D) Amounts of Gb3 and Lyso-Gb3 in the liver of Fabry mice transplanted with GLA-KO or GLA-KO+mNAGA iPSCs (n = 6). *P < 0.05, **P < 0.01, and NS (P > 0.1). mNAGA: modified α-N-acetylgalactosaminidase; iPSCs: induced pluripotent stem cells; GLA: galactosidase A; NS: not significantly different; Gb3: globotriaosylceramide; Lyso-Gb3: globotriaosylsphingosine.
To investigate whether transplanted GLA-KO+mNAGA iPSCs improved the GLA activity in these mice, we measured the GLA activity in the liver, heart, kidney, and blood plasma. We observed no recovery of the GLA activity in the heart, kidney, or blood plasma. However, the GLA activity in the liver was significantly improved by the transplantation of GLA-KO+mNAGA iPSCs (P = 0.0213; Fig. 4C). We also quantified the amounts of Gb3 and Lyso-Gb3 in the liver, but there was no detectable reduction of the substrates (Fig. 4D). Taken together, these data indicate that although further optimization is required, we delivered mNAGA from transplanted iPSCs to the liver in vivo.
Discussion
We generated human iPSCs expressing and secreting mNAGA by genome editing. Furthermore, the GLA gene was disrupted to exclude the potential immunogenicity of the endogenous GLA in iPSCs. We investigated the genomic sequences of 22 predicted off-target sites, and found no change between before and after the GLA disruption (Supplemental Fig. S1). Although more thorough analysis such as whole-genome sequencing should be performed for clinical applications, our results support that similar genome editing strategies can be applied to other disorders associated with the loss of functional molecules.
Co-culture experiments demonstrated that genetically engineered iPSCs could secrete mNAGA and restore the GLA activity by 24.7% and 41.8% in GLA-deficient iPS-fibroblasts and cardiomyocytes compared with WT cells, respectively. We also successfully established an immunodeficient Fabry mouse model. Then, we transplanted iPSCs into these mice to form teratomas. Although teratoma formation is not applicable as a treatment option, it was the best experimental approach, because teratoma formation can engraft a large number of cells and the transplanted cells can be easily identified in the mouse body. Multiple clinical studies to transplant iPSC-derived cells are ongoing, and transplantation methods are also rapidly evolving. We think that our results show potential benefits of replacing iPSCs currently used for transplantation by genome-edited iPSCs in the future. One of the organs most affected by Fabry disease is the heart. Therefore, one promising therapeutic option is to transplant cardiomyocyte sheets generated from iPSCs secreting mNAGA. Thus, one advantage of genetically modified iPSCs as a drug delivery system is its ability to provide therapeutic molecules directly to target organs and tissues by differentiating the cells into specific cell types.
GLA-KO+mNAGA iPSC–derived teratomas improved the GLA activity only in the liver of the treated Fabry model mice. We previously described that lysosomal enzymes such as GLA are incorporated into cells via cation-independent mannose 6-phosphate receptors and asialoglycoprotein receptors on the cell surface. These receptors are abundantly expressed in hepatic cells but not in heart or kidney cells 29 . Thus, the majority of lysosomal enzymes administered in ERT are incorporated into liver cells30,31. Our results are consistent with these previous reports (Fig. 4C).
Despite the improvement, the GLA activity in the liver of mice treated with GLA-KO+mNAGA iPSCs was still much lower (1.3 nmol/h/mg protein) than in the WT mouse liver (18.8 nmol/h/mg protein). Therefore, we should enhance the production and secretion of mNAGA from iPSCs. The GLA activity in the GLA-KO+mNAGA iPSCs lysate was six times higher than that of WT. However, the difference in the culture medium was only 1.8 times. These results suggest that most mNAGA was retained in the lysosomes, and not actively secreted 32 . Several genes are involved in regulating the transportation of lysosomal enzymes33–35. Thus, overexpression or disruption of these genes potentially enhances the secretion or uptake of mNAGA. Recently, it was reported that transplantation of engineered spheroids composed of mouse embryonic fibroblasts reduced the amount of Lyso-Gb3 in Fabry model mice 36 . Therefore, the comparison of the two strategies would be beneficial.
We previously reported that the GLA activity of classical Fabry patients with severe symptoms and later-onset Fabry patients with milder symptoms was about 0.3% and 3.4% compared with control subjects, respectively 37 . The fact that only about 3% difference in the GLA activity results in marked differences in the symptoms indicates that the required amount of mNAGA provided from iPSCs to have therapeutic effects might be small. Therefore, enhancement of mNAGA production and secretion is important for clinical purposes.
In summary, this study demonstrated that genome-edited iPSCs secreting therapeutic molecules could serve as not only a resource of cell transplantation but also a drug delivery system.
Supplemental Material
sj-docx-1-cll-10.1177_09636897231173734 – Supplemental material for In Vivo Delivery of Therapeutic Molecules by Transplantation of Genome-Edited Induced Pluripotent Stem Cells
Supplemental material, sj-docx-1-cll-10.1177_09636897231173734 for In Vivo Delivery of Therapeutic Molecules by Transplantation of Genome-Edited Induced Pluripotent Stem Cells by Ittetsu Nakajima, Takahiro Tsukimura, Terumi Ono, Tomoko Shiga, Hiroshi Shitara, Tadayasu Togawa, Hitoshi Sakuraba and Yuichiro Miyaoka in Cell Transplantation
Supplemental Material
sj-tiff-2-cll-10.1177_09636897231173734 – Supplemental material for In Vivo Delivery of Therapeutic Molecules by Transplantation of Genome-Edited Induced Pluripotent Stem Cells
Supplemental material, sj-tiff-2-cll-10.1177_09636897231173734 for In Vivo Delivery of Therapeutic Molecules by Transplantation of Genome-Edited Induced Pluripotent Stem Cells by Ittetsu Nakajima, Takahiro Tsukimura, Terumi Ono, Tomoko Shiga, Hiroshi Shitara, Tadayasu Togawa, Hitoshi Sakuraba and Yuichiro Miyaoka in Cell Transplantation
Supplemental Material
sj-tiff-3-cll-10.1177_09636897231173734 – Supplemental material for In Vivo Delivery of Therapeutic Molecules by Transplantation of Genome-Edited Induced Pluripotent Stem Cells
Supplemental material, sj-tiff-3-cll-10.1177_09636897231173734 for In Vivo Delivery of Therapeutic Molecules by Transplantation of Genome-Edited Induced Pluripotent Stem Cells by Ittetsu Nakajima, Takahiro Tsukimura, Terumi Ono, Tomoko Shiga, Hiroshi Shitara, Tadayasu Togawa, Hitoshi Sakuraba and Yuichiro Miyaoka in Cell Transplantation
Footnotes
Acknowledgements
We thank Drs Ikuo Kawashima (Laboratory of Biomembrane), Yoichi Tajima (Genome Dynamics Project), Yoshinobu Iguchi (Laboratory of Neuropathology), Shogo Nagata (Tokyo Medical and Dental University), and Yohei Hayashi (RIKEN BRC) for their technical assistance. We also thank all lab members for their helpful discussions.
Author Contributions
I.N. and Y.M. designed the experiments. Y.M. and H. Shitara generated genome-edited iPSCs and mouse zygotes, respectively. T.O. conducted the co-culture experiments. I.N. and T.O. transplanted iPSCs to mice. T. Tsukimura, T.S., and T. Togawa performed the mass spectrometry. I.N. conducted all other experiments. I.N. and Y.M. wrote the manuscript with help from other authors. Y.M. and H. Sakuraba supervised the projects.
Availability of Data and Materials
All data and materials described in this study are available upon request. The donor plasmid, AAV-CAGGS-mNAGA, is available from Addgene (#196448).
Ethical Approval
This study was approved by the Recombinant DNA Experiments Committee of Tokyo Metropolitan Institute of Medical Science. The approval number: #22-003.
Statement of Human and Animal Rights
This study was approved by the Ethics and Animal Experimentation Committees of Tokyo Metropolitan Institute of Medical Science. The approval numbers: #21-49, #22-016.
Statement of Informed Consent
Informed consent was obtained for the use of WTC11 iPSC line.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the Japan Society for the Promotion of Science (JSPS) Grant-in-Aid for Scientific Research (B) (grant number 20H03442), JSPS Grant-in-Aid for Challenging Research (Pioneering) (grant number 20K21409), Interstellar Initiative from AMED (grant number 20jm0610032h0001), Takeda Science Foundation Medical Research Grant, and iPS Academia Japan Research Grant to Y.M.
Supplemental Material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.