
Abstract
Hypoxic injury to the brain is very intricate under the control of biochemical reactions induced by various factors and mechanisms. Long non-coding RNAs (lncRNAs) have already been revealed to affect pathological processes in the nervous system of different degrees. This research aimed to investigate the mechanisms implicated in hypoxic brain injury. β-Asarone mitigated the decrease of cell viability, superoxide dismutase activity, and mitochondrial membrane potential, as well as the increase of cell apoptosis, lactate dehydrogenase release, malondialdehyde content, and reactive oxidative species production by cobalt chloride. LncRNA ribonuclease P RNA component H1 (RPPH1) was discovered to be highly expressed in hypoxia-induced PC12 cells, and β-Asarone addition led to a decline in RPPH1 expression. RPPH1 overexpression reversed the effect of β-Asarone on hypoxia-induced injury in PC12 cells. Furthermore, we proved that RPPH1 could sponge miR-542-3p. Subsequently, death effector domain containing 2 (DEDD2) was proven as the downstream gene of RPPH1/miR-542-3p axis. Eventually, the whole regulation mechanism of RPPH1/miR-542-3p/DEDD2 axis was testified through rescue assays. The impacts of β-Asarone on hypoxia-induced PC12 cells could be countervailed by RPPH1 augment, which was also discovered to be neutralized in response to miR-542-3p overexpression or DEDD2 depletion. These findings offered a novel perspective for understanding neuroprotection.
Introduction
Stroke, also named as cerebrovascular disease, is a widespread destructive neurological disease, characterized by high mortality and morbidity rates 1 . Stroke can result in hypoxic-ischemic injury to the brain, which is affected by multiple factors, in sequence or in combination, thus causing cell injuries 2 . β-Asarone, a major constituent of Acorus tatarinowii Schott, can cross the blood-brain barrier (BBB) easily and develop neuroprotective functions, such as protecting neuron against apoptosis3,4. Nevertheless, the impact of β-Asarone on hypoxia-activated cell injury was scarcely investigated.
Long non-coding RNAs (lncRNAs) have been confirmed as a novel class of transcripts (longer than 200 nucleotides) without the capability to encode protein5,6. It is learnt in numerous studies that lncRNAs are upstream modulators of gene expression to influence the cellular activities of multiple diseases, including cerebrovascular disease7–9. For example, lncRNA H19 hinders endothelial-mesenchymal transition in diabetic retinopathy 10 . LncRNA-N1LR facilitates neuroprotective property against ischemic stroke through inhibition of p53 phosphorylation 11 . LncRNA H19 provokes retinal ischemia/reperfusion injury through boosting microglial pyroptosis and neuronal death 12 . The investigations of lncRNAs in hypoxic-ischemic injury of the brain are still inadequate.
LncRNA ribonuclease P RNA component H1 (RPPH1) has been reported to play a disease-promoter role. Zhang and Tang 13 have reported that RPPH1 depletion exerts suppressive influence on cell proliferation and tumorigenesis in breast cancer by downregulating miR-122. Cai et al 14 have demonstrated that RPPH1-mediated CDC42 expression promotes the formation of hippocampal neuron dendritic spine via the competition with miR-330-5p. In addition, RPPH1 facilitates the mesangial cell inflammation and proliferation in diabetic nephropathy through the interaction with Gal-3 15 . The specified function of RPPH1 in hypoxia-induced cell injury remains to be interrogated.
Our research aimed to probe into the possible molecular mechanism of RPPH1 in hypoxia-induced injury, and the interaction of RPPH1 and β-Asarone and their functions in hypoxia-treated PC12 cells (established via pretreatment with cobalt chloride [CoCl2]) were also investigated, which might provide a novel perspective for understanding hypoxic brain injury.
Materials and Methods
Cell Culture
PC12 cells were purchased from Culture Collection of Chinese Academy of Science (Shanghai, China) and incubated under standard conditions (95% air, 37°C, 5% CO2). Cells were seeded in Dulbecco’s Modified Eagle’s Medium (DMEM; Gibco, Grand Island, NY, USA) containing fetal bovine serum (FBS; Gibco). CoCl2 (Sigma) of 600 μmol/l was added into the culture medium to provoke the chemical hypoxia of cells. As a fat-soluble substance with a small molecular weight, β-Asarone (2,4,5-trimethoxy-1-propenylbenzene) was obtained from Acorus calamus (Acoraceae). Hypoxia-induced PC12 cells for 24 h were treated by different concentrations of β-Asarone (20, 30, 45 μg/ml) with purity up to 99.55%. Human embryonic kidney 293T (HEK293T cells), procured from ATCC (Manassas, VA, USA), were cultured in DMEM (PM150210) supplemented with 10% FBS (164210-500) and 1% P/S (PB180120) in a humidified environment with 5% CO2 at 37°C.
Cell Transfection
Hypoxia-induced PC12 cells treated with β-Asarone were plated into six-well dishes and cultivated. Short hairpin RNAs (shRNAs) targeting DEDD2 (shDEDD2) and non-targeting plasmids (shNC), together with RPPH1 overexpression plasmids or empty pcDNA3.1 vector, were, respectively, synthesized by Purity Biotechnology (Wuhan, Hubei, China). Moreover, miR-542-3p mimics and NC mimics (the negative control) were synthesized by Purity Biotechnology as well. Transfection was performed via Lipofectamine 3000 (Invitrogen, Carlsbad, CA, USA) for 48 h.
qRT-PCR Assay
TRIzol reagent from Invitrogen was leveraged for the extraction of RNA, which was then reversely transcribed into complementary DNA (cDNA) with cDNA Reverse Transcription Kit (Invitrogen). Then, quantitative real-time polymerase chain reaction (qRT-PCR) was conducted with SYBR Green Real-Time PCR Kit (Thermo Fisher Scientific, Waltham, MA, USA) on the Bio-Rad CFX96 System (Thermo Fisher Scientific). The 2−ΔΔCT method was selected for transcript quantification. As the internal control, glyceraldehyde 3-phosphate dehydrogenase (GAPDH)/U1 was adopted.
CCK-8 Assay
PC12 cells (about 2 × 103) under different treatments were placed into each well of 96-well plates in DMEM (Gibco) and cultured at indicated time points. Ten microliters of CCK-8 solution (Dojindo, Tokyo, Japan) was added into each well and incubated for another 4 h. Finally, the absorbance at 450 nm was measured with a microplate reader (Bio-Tek, Winooski, VT, USA).
TUNEL Staining
Using the In Situ Cell Death Detection Kit (Roche, Mannheim, Germany), differentially treated PC12 cells were stained for nnheim, Germany), differentially treated PC12 cells were stained for assay. To visualize cell nuclei, cells after staining were counterstained with 4′,6-diamidino-2-phenylindol (DAPI; Thermo Fisher Scientific) solution under dark at room temperature for 5 min. A fluorescence microscope (Olympus, Tokyo, Japan) was used to capture the image of fluorescence.
Malondialdehyde, Superoxide Dismutase, and Lactate Dehydrogenase Assay
For the assessment of the malondialdehyde (MDA) content, its absorption in the wavelength (500 nm) was monitored through an ELISA reader (Abcam, Cambridge, CA, USA). The activity of superoxide dismutase (SOD) and the release of lactate dehydrogenase (LDH) were separately detected with the corresponding assay kit (Jiancheng Bioengineering Institute, Nanjing, China).
Mitochondrial Membrane Potential Assay
Prepared PC12 cells of different treatments were transferred into 25-cm2 culture flask containing 5 × 105 cells/ml of matrigel. After the addition of PBS (Sigma-Aldrich, Milan, Italy) and mitochondrial membrane potential (MMP) probe JC-1 (10 μmol/l) (Beyotime, Nantong, China), cells were incubated for 10 min in darkness, followed by labeling through the fluorescent. To obtain the ratio of red to green fluorescence intensity, cells were assessed through confocal fluorescence ratio imaging by laser scanning confocal microscope (LSCM) (Olympus).
Reactive Oxidative Species Assay
For this assay, 1.5 ml Eppendorf that contained 1 ml of PBS was used to suspend cells in the logarithmic growth phase. Then dichloro-dihydro-fluorescein diacetate (DCFH-DA) was supplemented to the cell suspension to achieve the final density at 10 μmol/l, which was subsequently centrifuged at 1,000 × g for 5 min. After that, the reactive oxidative species (ROS) level of cells was measured based on 15 μl of cell suspension dropped onto a slice. For this measurement, LSCM with an excitation wavelength at 488 nm and an emission wavelength at 525 nm was needed.
Subcellular Fractionation
By means of Nuclear and Cytoplasmic Extraction Reagents (Thermo Fisher Scientific), the fractions of cytoplasm and nucleus were isolated in hypoxia-activated PC12 cells. The levels of RPPH1, GAPDH (cytoplasmic control), and U1 (nuclear control) were evaluated via qRT-PCR.
Dual-Luciferase Reporter Assay
The wild-type or mutant binding sequence of miR-542-3p in RPPH1 or DEDD2 3′-untranslated region (UTR) was synthesized and subcloned into pmirGLO dual-luciferase vector to construct RPPH1-WT/MUT and DEDD2-WT/MUT (Promega, Madison, WI, USA). RPPH1-WT/MUT vector or DEDD2-WT/MUT vector was co-transfected with miR-542-3p mimics or NC mimics into hypoxia-induced PC12 cells and HEK293T cells. After 48 h, Dual-Luciferase Report Assay System (Promega) was applied for the measurement of luciferase activity in the indicated groups.
RNA Pull-Down Assay
Biotinylated RPPH1 probe, miR-542-3p-WT/MUT, and negative control were separately synthesized and termed as Bio-RPPH1, Bio-miR-542-3p-WT/MUT, and Bio-NC. After that, the biotinylated RNA was incubated overnight with cell lysate (Invitrogen). RNA-bound beads (Millipore) adhered to streptavidin (Invitrogen) were incubated under the above conditions. Finally, purified RNA compound was assessed with qRT-PCR.
RNA Immunoprecipitation Assay
Through an Imprint RNA Immunoprecipitation (RIP) Kit (Sigma-Aldrich), RIP assay was conducted. The hypoxia-induced PC12 cells were harvested and then lysed in RIP lysis buffer (Invitrogen). After being added to magnetic beads (Millipore), the whole cell lysate was then incubated with negative control anti-IgG (Abcam) or anti-Ago2 (Abcam). The immunoprecipitated RNA was subjected to qRT-PCR analysis after the isolation and purification processes.
Western Blot Analysis
The complete proteome was lysed through RIPA lysis buffer (Invitrogen). After that, the BCA Protein detection kits (Thermo Fisher Scientific) were adopted to quantify proteins. After being separated through sodium dodecyl-sulfate polyacrylamide gel electrophoresis (SDS-PAGE), proteins were then blotted onto polyvinylidene difluoride (PVDF) membranes (Millipore, Billerica, MA, USA). The membranes were treated by 5% skim milk, followed by cultivation with specific primary antibodies (Abcam) including anti-MFN2 (ab56889), anti-SPRED1 (ab77079), anti-TCF12 (ab245540), anti-DEDD2 (ab36858), anti-PLPBP (ab251898), and anti-GAPDH (ab8245) at 4°C for one night, and further 1-h incubation with secondary antibody at 37°C. GAPDH served as the loading control. Protein bands were visualized via ECL (Pierce, Rockford, IL, USA).
Statistical Analysis
Results were assessed through GraphPad Prism 6.01 (GraphPad Software, Inc., La Jolla, CA, USA). Data were shown as means ± SD. Student’s t-test or analysis of variance (ANOVA) was performed to assay the differences. P < 0.05 had statistical significance. Each sample was evaluated in triplicate.
Results
β-Asarone Protected PC12 Cells Against Hypoxia-Induced Injury
PC12 cells were treated with CoCl2 for the induction of hypoxia. Cell viability was weakened in a time-dependent manner after the exposure to hypoxia (0, 6, 12, and 24 h) (Fig. 1A). Finally, 24-h hypoxia treatment was chosen for the following experiments. The chemical structure of β-Asarone was demonstrated (Fig. 1B). Then, various doses of β-Asarone were added to treat PC12 cells after 24-h hypoxia treatment. Obviously, the treatment with 30 μg/ml of β-Asarone rescued the inhibited cell viability induced by hypoxia to the largest extent (Fig. 1C). Thus, β-Asarone (30 μg/ml) was selected as the optimal treatment in our study. Besides, TUNEL assay illustrated that apoptosis of PC12 cells promoted by hypoxia was repressed by the addition of β-Asarone (Fig. 1D). Compared with PC12 cells in the control group, the increase in LDH and MDA contents and the decrease in SOD activity were observed in the hypoxia group, which were reversed by β-Asarone treatment (Fig. 1E–G). In addition, JC-1 assay showed that hypoxia-decreased MMP was recovered when β-Asarone was added to PC12 cells (Fig. 1H). Higher ROS level in the hypoxia group was reduced by the addition of β-Asarone (Fig. 1I). After these detections, we concluded that β-Asarone played a protective role in hypoxia-induced PC12 cell injury.

β-Asarone protected PC12 cells against hypoxia-induced injury. (A) CCK-8 assay was done for the evaluation of PC12 cell viability under the influence of hypoxia treatment at various time points. (B) The chemical structural formula of β-Asarone was displayed. (C) The reduced cell viability of hypoxia-treated PC12 cells was rescued by β-Asarone addition. (D) TUNEL assay was done for the evaluation of PC12 cells treated with hypoxia and β-Asarone. (E-F) The LDH release and MDA content of PC12 cells were measured under the conditions of hypoxia treatment and the addition of β-Asarone. (G) The inhibition of SOD activity in PC12 cells, resulting from hypoxia treatment, was enhanced through β-Asarone. (H) β-Asarone strengthened MMP in hypoxia-treated PC12 cells. (I) β-Asarone inhibited ROS production of hypoxia-treated PC12 cells. DAPI: 4′,6-diamidino-2-phenylindol; LDH: lactate dehydrogenase; MDA: malondialdehyde; MMP: mitochondrial membrane potential; ROS: reactive oxidative species; SOD: superoxide dismutase. *P < 0.05; **P < 0.01.
RPPH1 Overexpression Neutralized the Impact of β-Asarone on Hypoxia-Stimulated PC12 Cells
LncRNAs are known to play pivotal roles during the pathological processes of cells16–18. Therefore, we sought to explore the possible lncRNA implicated in hypoxia-induced PC12 cells. LncRNA RPPH1 has been identified in assorted human cancers and diseases to exert the regulatory function13–15; thus, we aimed to fathom out its function in hypoxia-induced PC12 cell injury. The expression of RPPH1 in control, hypoxia, and hypoxia + β-Asarone groups was examined through qRT-PCR, respectively. In PC12 cells, the increment in RPPH1 expression under the influence of hypoxia treatment was recovered by the addition of β-Asarone (Fig. 2A). Subsequently, the decline in RPPH1 level as a result of β-Asarone treatment in hypoxia-induced PC12 cells was increased by the transfection of RPPH1 overexpression vectors (Fig. 2B). CCK-8 demonstrated that β-Asarone promoted the viability of hypoxia-induced PC12, and this effect was then countervailed by RPPH1 overexpression (Fig. 2C). TUNEL assay indicated that β-Asarone restrained apoptosis of hypoxia-induced PC12 cells, while this phenomenon was reversed on account of RPPH1 overexpression (Fig. 2D). Moreover, the decreased LDH and MDA contents and elevated SOD activity caused by β-Asarone treatment were reversed on account of RPPH1 upregulation (Fig. 2E–G). In addition, JC-1 experiment exhibited that MMP elevated with β-Asarone addition was reduced by RPPH1 overexpression (Fig. 2H). Also, β-Asarone lessened ROS level, and the trend was recovered after the upregulation of RPPH1 (Fig. 2I). Totally, β-Asarone protects PC12 cells against hypoxia-induced injury via downregulating RPPH1.

RPPH1 overexpression reversed the effect of β-Asarone on hypoxia-induced injury in PC12 cells. (A) RPPH1 was upregulated in hypoxia-treated PC12 cells, but its expression was reduced by β-Asarone. (B) The expression of RPPH1 was calculated via qRT-PCR in hypoxia-induced PC12 cells under conditions of β-Asarone treatment and transfection with RPPH1 overexpression vector. (C) The viability of hypoxia-induced PC12 cells was evaluated under the influence of β-Asarone treatment and RPPH1 upregulation. (D–F) The decrease in apoptosis, LDH release, and MDA content of hypoxia-treated PC12 cells induced by β-Asarone was ameliorated by RPPH1 overexpression. (G–H) The SOD activity and MMP of hypoxia-induced PC12 cells were detected after β-Asarone treatment and RPPH1 overexpression. (I) RPPH1 upregulation could enhance the decreased ROS level in β-Asarone-added PC12 cells under hypoxia. DAPI: 4′,6-diamidino-2-phenylindol; LDH: lactate dehydrogenase; MDA: malondialdehyde; MMP: mitochondrial membrane potential; qRT-PCR: quantitative real-time polymerase chain reaction; RPPH1: ribonuclease P RNA component H1; ROS: reactive oxidative species; SOD: superoxide dismutase. **P < 0.01.
RPPH1 Targeted MiR-542-3p in Hypoxia-Treated PC12 Cells
The regulation mechanism underlying lncRNAs largely depends on the position of lncRNAs in cells19,20. With GAPDH and U1 as the cytoplasmic and nuclear controls, respectively, subcellular fractionation assays were applied for confirmation of RPPH1 distribution in cells, and it was discovered that a large proportion of RPPH1 was located in the cytoplasm of hypoxia-treated PC12 cells, indicating the regulation at posttranscriptional level (Fig. 3A). Hence, the competing endogenous RNA (ceRNA) network implicated in the regulation mechanism of various lncRNAs was taken into consideration. Then, based on prediction from starBase (http://starbase.sysu.edu.cn/index.php), seven microRNAs (miRNAs) were discovered to bind with RPPH1 under indicated conditions (CLIP-Data ≥5; pan-Cancer ≥2). For further screening, we implemented RNA pull-down assay and discovered that miR-542-3p and miR-328-3p were significantly pulled down by the biotinylated RPPH1 probe (Bio-RPPH1) relative to the control group (Bio-NC), indicating the potential binding correlation between the two miRNAs and RPPH1 (Fig. 3B). Furthermore, we measured the expression of miR-328-3p and miR-542-3p in PC12 cells under the conditions of hypoxia and β-Asarone treatment. qRT-PCR results found that the decline in miR-328-3p expression induced by hypoxia cannot be reversed by β-Asarone, while the reduction in miR-542-3p expression caused by hypoxia was reversed as a result of β-Asarone supplement (Fig. 3C). Then we overexpressed miR-542-3p in cells by transfecting miR-542-3p mimics, with NC mimics serving as the control (Fig. 3D). To confirm the binding between RPPH1 and miR-542-3p, we carried out dual-luciferase reporter assay and RNA pull-down assay. The binding sites between RPPH1 and miR-542-3p are presented in Fig. 3E. In addition to hypoxia-induced PC12 cells, dual-luciferase reporter assays were also conducted in HEK293T cells for further confirmation as this cell line (HEK293T) was very easy to grow and to maintain, with high reproducibility, which was frequently used in many experiments, including luciferase reporter assay 21 . As we expected, relative to the control group (NC mimics), the transfection of miR-542-3p mimics resulted in an overt decline in the luciferase activity of RPPH1-WT, while no obvious difference in that of RPPH1-MUT was discovered in cells with or without miR-542-3p overexpression (Fig. 3F–G). In RNA pull-down assay, RPPH1 was abundantly precipitated in the Bio-miR-542-3p-WT group rather than in Bio-miR-542-3p-MUT or Bio-NC (the control group) (Fig. 3H). These data displayed that RPPH1 could bind to miR-542-3p in hypoxia-induced PC12 cells.
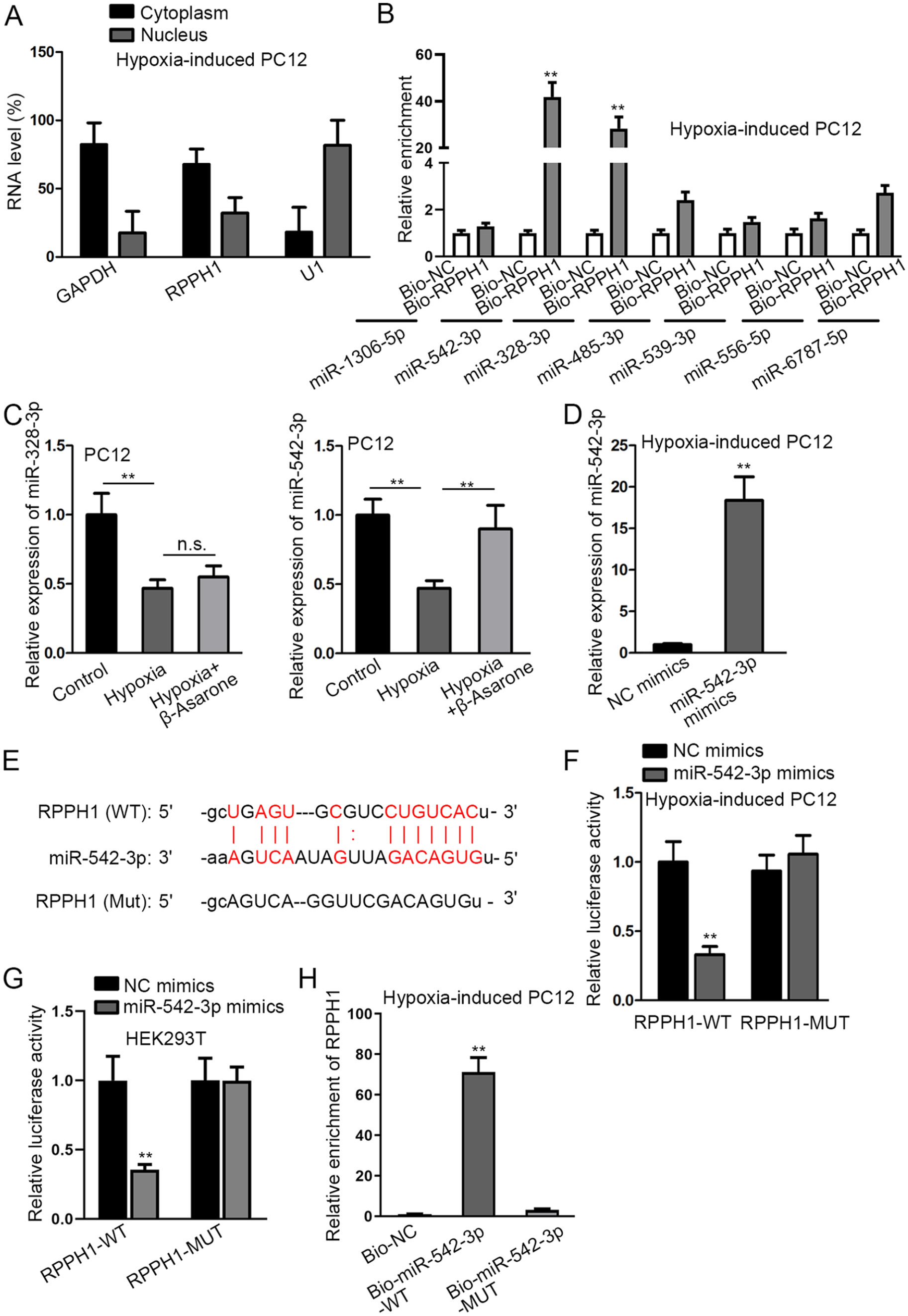
RPPH1 could bind to miR-542-3p. (A) With GAPDH and U1 as the cytoplasmic and nuclear controls, respectively, the distribution of RPPH1 was determined through subcellular fractionation assay. (B) RNA pull-down assay was done for the screening of the candidate miRNA binding to RPPH1 by detecting the miRNA levels pulled down by biotin-labeled RPPH1 (Bio-RPPH1) and the control group (Bio-NC) in hypoxia-treated PC12 cells. (C) The expression of miR-328-3p and miR-542-3p was detected via qRT-PCR in hypoxia-treated PC12 cells with or without β-Asarone addition. (D) qRT-PCR analysis was done for the measurement of miR-542-3p expression in hypoxia-treated PC12 cells in response to the transfection of miR-542-3p mimics, with NC mimics as the negative control, whose transfection cannot result in an overt increase in miR-542-3p expression. (E) The binding sites of miR-542-3p and RPPH1 were demonstrated. (F–G) The binding sites between RPPH1 and miR-542-3p were confirmed by dual-luciferase reporter assay in cells with or without miR-542-3p upregulation (through the transfection with miR-542-3p mimics or NC mimics). In addition to hypoxia-treated PC12 cells, dual-luciferase reporter assay was also conducted in HEK293T for further confirmation, which was very easy to grow and to maintain, with high reproducibility, and frequently used in various experiments. (H) The binding of RPPH1 and miR-542-3p was further proved by RNA pull-down assay based on the measurement of RPPH1 enrichment in Bio-miR-542-3p-WT/MUT and the control group (Bio-NC). GAPDH: glyceraldehyde 3-phosphate dehydrogenase; HEK293T: human embryonic kidney 293T; NC: negative control; n.s.: no significance; qRT-PCR: quantitative real-time polymerase chain reaction; RPPH1: ribonuclease P RNA component H1. **P < 0.01.
DEDD2 Was Discovered as the Downstream Target of RPPH1/MiR-542-3p
To find the downstream genes, we searched on starBase and finally obtained five common messenger RNAs (mRNAs) targeted by miR-542-3p, namely, MFN2, SPRED1, DEDD2, TCF12, and PLPBP (Fig. 4A). The effects of miR-542-3p on these mRNAs were estimated via qRT-PCR and Western blot. The results demonstrated that death effector domain containing 2 (DEDD2) mRNA and protein levels were significantly decreased when miR-542-3p was overexpressed, whereas no obvious variations were discovered in the expression levels of the other four mRNAs (Fig. 4B, C). The expression of DEDD2 in PC12 cells under different treatments was then measured. Based on the results, the augment in DEDD2 expression, resulting from hypoxia treatment, was discovered to be reversed in response to β-Asarone addition (Fig. 4D). Besides, the mRNA and protein levels of DEDD2 were augmented in hypoxia-treated PC12 cells with RPPH1 upregulation (Fig. 4E). Sequentially, the interaction between miR-542-3p and DEDD2 was investigated. The binding sequences of the 3′-UTR of DEDD2 for miR-542-3p and the corresponding mutated sites are shown in Fig. 4F. Dual-luciferase reporter assays were then performed in hypoxia-induced PC12 and also HEK293T cells for the verification of the binding sites. According to the experimental results, the luciferase activity of DEDD2-WT was overtly inhibited under the influence of miR-542-3p mimics transfection, while the luciferase activity of DEDD2-MUT was hardly affected under the same conditions (Fig. 4G, H). In RNA pull-down experiments, as compared with the control group (Bio-NC) and Bio-miR-542-3p-MUT, DEDD2 was more abundantly pulled down by Bio-miR-542-3p-WT probe (Fig. 4I). In addition, based on RIP results, RPPH1, miR-542-3p, and DEDD2 were all largely precipitated in the Ago2 group rather than the negative control (IgG group), hinting the coexistence of RPPH1, miR-542-3p, and DEDD2 in RNA-inducing silencing complex (RISC) (Fig. 4J). The findings together suggested that DEDD2 was targeted by RPPH1/miR-542-3p.

DEDD2 was the downstream target gene of miR-542-3p. (A) The predicted number of mRNAs of miR-542-3p was shown in Venn diagram. (B–C) The mRNA and protein levels of candidate 5 mRNAs were measured by qRT-PCR and Western blot (WB) in hypoxia-treated PC12 cells with or without miR-542-3p overexpression under the influence of the transfection with miR-542-3p mimics or NC mimics. (D) An overt increment in the mRNA and protein levels of DEDD2 in hypoxia-treated PC12 cells was reversed by β-Asarone treatment. (E) qRT-PCR and WB analyses were done for the measurement of DEDD2 expression under the influence of RPPH1 upregulation. (F–G) After the acquisition of potential binding site between miR-542-3p and DEDD2, dual-luciferase reporter assay was done for verification of the combination between miR-542-3p and DEDD2 by detecting the luciferase activity of DEDD2-WT/MUT in hypoxia-treated PC12 cells with or without miR-542-3p overexpression under the influence of the transfection with miR-542-3p mimics or NC mimics. (H) For further confirmation, dual-luciferase reporter assay was also conducted in HEK293T, which was frequently used in various experiments for their ease of transfection and culture with high reproducibility. (I) The combination of miR-542-3p and DEDD2 was verified via RNA pull-down assays by measuring the enrichment of DEDD2 in Bio-miR-542-3p-WT/MUT and the control group (Bio-NC). (J) The enrichment of RPPH1, miR-542-3p, and DEDD2 in the Ago2 and IgG group (the negative control) was measured in RIP assay. DEDD2: death effector domain containing 2; GAPDH: glyceraldehyde 3-phosphate dehydrogenase; HEK293T: human embryonic kidney 293T; mRNA: messenger RNA; NC: negative control; qRT-PCR: quantitative real-time polymerase chain reaction; RIP: RNA Immunoprecipitation; RPPH1: ribonuclease P RNA component H1. **P < 0.01.
Impacts of RPPH1 Overexpression on Hypoxia-Induced PC12 Cells With β-Asarone Treatment Could Be Counteracted by MiR-542-3p Upregulation or DEDD2 Downregulation
With the aim to validate the function of RPPH1/miR-542-3p/DEDD2 axis in hypoxia-induced PC12 cells, rescue assays were performed under the treatment of β-Asarone and transfection with indicated plasmids. DEDD2 knockdown efficiency was first determined (Fig. 5A). As was illustrated in the above experiments, β-Asarone exerted protective function in hypoxia-induced PC12 cells. Based on CCK-8 assay, the strengthened viability of hypoxia-induced PC12 cells as a result of β-Asarone treatment was weakened by RPPH1 upregulation, which could be rescued by miR-542-3p overexpression or DEDD2 silence (Fig. 5B). TUNEL assay indicated that the overt decline in apoptosis rate induced by β-Asarone addition was reversed in response to RPPH1 overexpression, whose effect was then countervailed by upregulating miR-542-3p or silencing of DEDD2 (Fig. 5C). Furthermore, the repressed LDH and MDA contents and increased SOD activity, resulting from β-Asarone addition, were reversed by RPPH1 overexpression, and miR-542-3p overexpression or DEDD2 knockdown counteracted the effect of RPPH1 overexpression (Fig. 5D–F). In addition, the promoting impact of β-Asarone treatment on MMP was neutralized by the augment of RPPH1, which was later inversely changed in response to miR-542-3p upregulation or DEDD2 repression (Fig. 5G). Moreover, the decrease in ROS level induced by β-Asarone addition was notably reversed in response to RPPH1 overexpression and was again reduced by miR-542-3p mimics or shDEDD2 transfection (Fig. 5H). In conclusion, β-Asarone regulated hypoxia-activated injury in PC12 cells through modulating RPPH1/miR-542-3p/DEDD2 axis.

The effect of RPPH1 overexpression on hypoxia-induced PC12 cells could be countervailed by miR-542-3p overexpression or DEDD2 depletion. (A) The mRNA and protein levels of DEDD2 were both reduced by shDEDD2 transfection in hypoxia-treated PC12 cells. Rescue assays were then implemented in hypoxia-induced PC12 cells treated with or without β-Asarone and transfected with indicated plasmids including pcDNA3.1, RPPH1 overexpression vector, NC mimics and miR-542-3p mimics, shDEDD2, and the shNC. (B) The increased viability of hypoxia-induced PC12 cells, resulting from β-Asarone treatment, was inhibited by RPPH1 upregulation, which was then reversed in response to augmented miR-542-3p (transfection with miR-542-3p mimics) or shDEDD2. (C–E) The decline in apoptosis rate, LDH content, and MDA content of hypoxia-induced PC12 cells as a result of β-Asarone treatment was enhanced on account of an increment in RPPH1 level, which was reversed by overexpressing miR-542-3p or silencing DEDD2. (F–G) SOD activity and MMP of hypoxia-induced PC12 cells were evaluated in response to β-Asarone treatment and transfection with indicated plasmids. (H) ROS production of hypoxia-treated PC12 cells was detected in indicated groups. DAPI: 4′,6-diamidino-2-phenylindol; DEDD2: death effector domain containing 2; LDH: lactate dehydrogenase; MDA: malondialdehyde; MMP: mitochondrial membrane potential; mRNA: messenger RNA; NC: negative control; RPPH1: ribonuclease P RNA component H1; ROS: Reactive Oxidative Species; shDEDD2: silenced DEDD2; shNC: corresponding negative control; SOD: superoxide dismutase. **P < 0.01.
Discussion
Recent reports have indicated the function of lncRNAs in different kinds of cell injuries, especially in PC12 cells. Highly expressed lncRNA ATB enhances amyloid-β-activated neurotoxicity of PC12 cells via regulation of miR-200/ZNF217 pathway 22 . LncRNA CCAT1 regulates neuropathic pain by serving as a sponge of miR-155 16 . LncRNA ROS is ascertained to facilitate the cerebral hypoxia/reoxygenation injury in PC12 cells via modulation on miR-135a-5p and ROCK1/2 18 . As the pathomechanism of cell injury is extremely complicated, more efforts are needed for the discovery of novel effective biomarkers.
PC12 injury models were constructed via treatment of CoCl2. Then β-Asarone was added to observe the protective role of β-Asarone in PC12 cells. β-Asarone was validated to countervail the effect of hypoxia on PC12 cells due to the changes in cell viability, cell apoptosis, LDH and MDA contents, SOD activity, MMP, and ROS level. To find out the feasible mechanism in hypoxia-stimulated PC12 cells under β-Asarone treatment, we focused on the correlation between lncRNA RPPH1 and β-Asarone. Past studies have revealed the promoting function of RPPH1 in diseases13,14,23. Function assays certified that RPPH1 upregulation reversed the effects of addition of β-Asarone on hypoxia-induced PC12 cells. This research, for the first time, raveled out the function of RPPH1 in hypoxia-induced injury in PC12 cells.
Based on the cytoplasmic location of RPPH1 in hypoxia-treated PC12 cells, we suspected that RPPH1 might function via ceRNA network. In other words, RPPH1 could bind to certain miRNA to modulate the expression of the downstream target mRNA, thus exerting its impacts on hypoxia-induced PC12 cells. According to the prediction on starBase v2.0 and results of mechanism experiments, miR-542-3p was finally proved as the miRNA binding to RPPH1. Besides, numerous research works have indicated the regulatory role of miR-542-3p in other diseases. For instance, miR-542-3p targeting survivin overcomes HER3 signaling–promoted chemoresistance and enhances the tumor-repressive activity of paclitaxel against breast cancer with HER2 elevation 24 . Tumor cell–secreted angiogenin boosts the angiogenic process of endothelial cells via suppression of miR-542-3p 25 . MiR-542-3p contributes to cell apoptosis in dermal fibroblasts collected from systemic sclerosis patients via targeting survivin 26 . Sequentially, DEDD2 was chased down as the downstream gene of RPPH1/miR-542-3p axis by means of bioinformatics analyses and mechanism experiments. DEDD2 is expressed widely in human tissues with high level, and DEDD2 upregulation has been proven to induce moderate apoptosis. In the current study, RPPH1 was discovered to upregulate DEDD2 via binding to miR-542-3p. In addition, it was further validated that the protective impacts of β-Asarone on hypoxia-treated PC12 cells were countervailed in response to RPPH1 overexpression, and this effect was discovered to be neutralized by miR-542-3p overexpression or DEDD2 silence. Thus, the function of RPPH1/miR-542-3p/DEDD2 axis in PC12 cells induced by hypoxia was ascertained.
Along these lines, RPPH1 was first found to bind with miR-542-3p to upregulate DEDD2 expression, thus enhancing the hypoxia injury of PC12 cells. Moreover, the protective role of β-Asarone in hypoxia-stimulated PC12 cells depended on its suppression on RPPH1/miR-542-3p/DEDD2 axis. In summary, a novel perspective could be developed for better understanding of hypoxic-ischemic brain injury.
Footnotes
Acknowledgements
We appreciate the supports of our experimenters.
Ethical Approval
Not applicable.
Statement of Human and Animal Rights
Not applicable.
Statement of Informed Consent
Not applicable.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.