
Abstract
Myocardial infarction (MI) is a severe life-threatening disease caused by acute and persistent ischemia and hypoxia and eventually leads to heart failure and sudden death. Long noncoding RNAs (lncRNAs) play significant roles in the pathology, diagnosis, and development of various cardiovascular diseases, including MI. This study aimed to explore the effect and molecular mechanism of lncRNA miR-22 host gene (MIR22HG) on hypoxia-induced injury in AC16 cardiomyocytes. The expression of MIR22HG and miR-24 in hypoxia-treated AC16 cardiomyocytes was detected by quantitative real-time polymerase chain reaction. Cell viability, lactate dehydrogenase release, levels of aspartate aminotransferase (AST) and creatine kinase-MB (CK-MB), and apoptosis were detected by Cell Counting Kit-8, lactate dehydrogenase (LDH) release assay, commercial enzyme-linked immune sorbent assay kits, and flow cytometry analysis, respectively. The protein levels of nuclear factor-kappa B (NF-κB) p65 and cytoplasmic inhibitor of kappa B alpha (IκBα) and phosphorylated IκBα were detected by western blot. Results showed that hypoxia treatment decreased viability and increased MIR22HG expression in AC16 cardiomyocytes. MIR22HG overexpression aggravated hypoxia-induced viability reduction, leakage of myocardial injury markers LDH, AST, and CK-MB, and apoptosis in AC16 cardiomyocytes, while MIR22HG knockdown elicited the reverse effects. MIR22HG overexpression enhanced NF-κB activation in hypoxia-treated AC16 cardiomyocytes. Inhibition of NF-κB pathway impaired the effects of MIR22HG overexpression on hypoxia-induced injury in AC16 cardiomyocytes. Moreover, MIR22HG knockdown inhibited the NF-κB pathway by upregulating miR-24 in AC16 cardiomyocytes. Inhibition of miR-24 resisted the effects of MIR22HG silencing on hypoxia-induced injury in AC16 cardiomyocytes. In conclusion, MIR22HG overexpression aggravated hypoxia-induced injury in AC16 cardiomyocytes via enhancing NF-κB activation by targeting miR-24.
Introduction
Coronary heart disease (CHD), also known as ischemic heart disease, is one of the leading causes of heart disease-related morbidity and mortality in both developing and developed countries 1 . CHD thus poses an enormous threat to human health worldwide, particularly in the middle-aged and elderly population 2 . As the most serious manifestation of CHD, myocardial infarction (MI) is a severe life-threatening disease caused by acute and persistent ischemia and hypoxia due to occlusion of the coronary artery, leading to an irreversible cardiomyocyte loss and cardiac dysfunction 3 . Severe and sustained pain in the chest complicating with malignant arrhythmia, fever, syncope, and ultimately heart failure is the major clinical symptom for patients with MI 4,5 . Hypoxia mainly results from myocardial ischemia and causes cardiomyocyte apoptosis or death and progressive injury to cardiac tissues 6,7 , leading to pathological cardiac remodeling and ultimately heart failure 8,9 . Despite substantial progress in myocardial protection research, many people still eventually succumb to this disease 10,11 . Therefore, the development of approaches to alleviate hypoxia-induced cardiomyocyte injury has recently emerged as a crucial avenue of research for the treatment of patients with MI.
Long noncoding RNAs (lncRNAs) are known as a novel class of highly conserved endogenous noncoding transcripts longer than 200 nucleotides with no or limited protein-coding function 12 . It has been well documented that lncRNAs are emerging as critical players in various physiological and pathological processes, such as cell proliferation, differentiation, inflammation, apoptosis, and metabolism 13,14 . To date, there is increasing evidence that lncRNAs are aberrant expressed and play significant roles in the pathology, diagnosis, and development of various cardiovascular diseases, including MI 15,16 . In this regard, identifying MI-associated lncRNAs and deciphering their related molecular mechanisms may be helpful to open up a new avenue for molecular therapeutics of MI.
LncRNA NR_028502.1, located at chromosome 17p13.3, is annotated as the human miR-22 host gene (MIR22HG). MIR22HG, mainly exists in the cytoplasm, was firstly identified as surrogate indicators of model chemical stress response in human-induced pluripotent stem cells 17,18 . MIR22HG has been demonstrated to be frequently deleted, hypermethylated, or exhibits loss of heterozygosity in hepatocellular carcinoma 19 . Notably, Li et al. reported that MIR22HG was overexpressed in acute myocardial infarction (AMI) samples compared with that in healthy samples according to the lncRNA expression analysis using the Gene Expression Omnibus database GSE66360 and GSE48060 20 . Meanwhile, Zangrando et al. also provided the evidence that a higher expression of MIR22HG was identified in the heart tissues of MI mice compared to that in sham-operated mice using significance analysis of microarrays method under GSE46395 21 . However, the detailed role and mechanism of MIR22HG in MI remain largely unknown. In the present study, we proved that hypoxia increased MIR22HG expression in AC16 cardiomyocytes. Moreover, MIR22HG overexpression aggravated hypoxia-induced injury in AC16 cardiomyocytes by enhancing nuclear factor-kappa B (NF-κB) activation via targeting miR-24-3p.
Materials and Methods
Cell Treatment
Human AC16 cardiomyocytes, obtained from the ATCC (Manassas, VA, USA), were maintained in Dulbecco’s modified Eagle’s medium (Gibco, Grand Island, NY, USA) with the existence of 10% fetal bovine serum (HyClone, South Logan, UT, USA), 100 U/ml penicillin, and 100 μg/ml streptomycin (Thermo Fisher Scientific, Waltham, MA, USA) at 37°C in a humidified atmosphere containing 5% CO2 and 95% air. To induce hypoxic injury of AC16 cardiomyocytes, AC16 cardiomyocytes were exposed to hypoxia in a modular incubator (Thermo Fisher Scientific) with 94% N2, 5% CO2, and 1% O2 for 12, 24, 36, and 48 h at 37°C. AC16 cardiomyocytes in the normoxic control group were cultured in a normoxic incubator with 95% air and 5% CO2 at 37°C.
Cell Transfection
MIR22HG-overexpressing plasmid pcDNA-MIR22HG (termed MIR22HG) and its corresponding control (termed Vector), small interfering RNAs targeting MIR22HG (termed si-MIR22HG-1 and si-MIR22HG-2) and their scramble control (termed Scramble), and miR-24 inhibitor (termed anti-miR-24) and inhibitor control (termed anti-miR-con) were designed and synthesized by RiboBio Co., Ltd (Guangzhou, China). Transient transfection of AC16 cardiomyocytes with oligonucleotides or plasmids was carried out using Lipofectamine 3000 reagent (Invitrogen, Carlsbad, CA, USA).
Quantitative Real-time Polymerase Chain Reaction
A RNeasy Mini Kit (QIAGEN, Dusseldorf, Germany) was utilized to isolate total RNA from treated AC16 cardiomyocytes. Reverse transcription was carried out to synthesize the first-strand cDNA using the QuantiTect Reverse Transcription Kit (Qiagen, Valencia, CA, USA). Thereafter, qPCR was performed to detect MIR22HG and miR-24 expression using SYBR® Premix Ex Taq™ reagent (TaKaRa, Shiga, Japan) and Taqman Universal Master Mix II (Applied Biosystems, Foster City, CA, USA) on a QuantStudio 3 Real-Time PCR System (Thermo Fisher Scientific), respectively. The relative gene expression was quantified using 2−ΔΔCt method. Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) and U6 were used as the normalization for MIR22HG and miR-24, respectively. The primer sequences were: MIR22HG, forward 5′-CGG ACG CAG TGA TTT GCT-3′ and reverse 5′-GCT TTA GCT GGG TCA GGA CA-3′; GAPDH, forward 5′-GGA GCG AGA TCC CTC CAA AAT-3′ and reverse 5′-GGC TGT TGT CAT ACT TCT CAT GG-3′; miR-24, forward 5′-TGG CTC AGT TCA GCA GGA ACA G-3′ and reverse 5′-GCG GTC ATC GAC ACG CTA CGC AAC G-3′; and U6, forward 5′-CTC GCT TCG GCA GCA CA-3′ and reverse 5′-AAC GCT TCA CGA ATT TGC GT-3′.
Cell Counting Kit-8 Assay
Cell viability was measured using Cell Counting Kit-8 (CCK-8; Dojindo, Laboratories, Kumamoto, Japan) according to the manufacturer’s instructions. To be more specific, AC16 cardiomyocytes were plated into the 96-well plates at a density of 10,000 cells per well and exposed to hypoxia or normoxia for 12, 24, 36, and 48 h. Sometimes, AC16 cells were transfected with MIR22HG, si-MIR22HG, si-MIR22HG plus anti-miR-24, si-MIR22HG plus anti-miR-con, or corresponding controls and then treated with 10 μM pyrrolidine dithiocarbamate (PDTC) (Sigma, St Louis, MO, USA), an inhibitor of NF-κB signaling, prior to hypoxia exposure for 36 h. After the indicated treatments, 10 µl of CCK-8 solution was supplemented into each well for another 4 h of incubation at 37°C. Finally, the optical density at a wavelength of 450 nm was recorded by a Microplate Reader (Bio-Rad, Hercules, CA, USA).
Lactate Dehydrogenase Release Assay
The release of lactate dehydrogenase (LDH) in the culture supernatant was regarded as an indicator of cellular injury. After the indicated treatments, treated AC16 cardiomyocytes were incubated with 1% Triton X-100 lysing solution (Sigma) for 20 min and the culture supernatant was collected by centrifugation. LDH level in the supernatant was detected using the LDH Cytotoxicity Assay Kit (Beyotime Institute of Biotechnology, Nanjing, China) referring to the manufacturer’s instructions. The absorbance at 450 nm was recorded by means of a Microplate Reader (Bio-Rad).
Measurement of Aspartate Aminotransferase and Creatine Kinase-MB Levels
The levels of myocardial-specific marker aspartate aminotransferase (AST) and creatine kinase-MB (CK-MB) in the cell supernatants were analyzed using respective commercial enzyme-linked immune sorbent assay (ELISA) kits (Jiancheng Bioengineering Institute, Nanjing, China) referring to the manufacturer’s instructions.
Flow Cytometry Analysis
An Annexin V-fluorescein isothiocyanate (FITC) Apoptosis Detection kit (BD Biosciences, San Jose, CA, USA) was employed to detect the percentage of apoptotic cells. After the indicated treatments, AC16 cardiomyocytes were collected by trypsinization and gently washed with ice-cold phosphate buffered saline (Bio-Rad). Afterward, AC16 cardiomyocytes were resuspended in 200 µl binding buffer containing 5 µl FITC-annexin V and 5 µl propidium iodide. After incubation at room temperature for 15 min in the dark, the apoptotic cells were analyzed by a flow cytometer (FACScan; BD Biosciences).
Western Blot Analysis
Cytoplasmic and nuclear proteins of treated AC16 cardiomyocytes were isolated using the ProteoJET Cytoplasmic and Nuclear Protein Extraction Kit (Fermentas, Glen Burnie, MD, USA), respectively. Equivalent amount of protein samples were loaded on 10% sodium dodecyl sulfate polyacrylamide gel electrophoresis for separation and then electrotransferred onto polyvinylidene fluoride membranes. After immersed in 5% nonfat milk for 1 h to block nonspecific sites, the membranes were incubated with the primary antibodies including NF-κB p65, inhibitor of kappa B alpha (IκBα), phosphorylated IκBα (p-IκBα), Lamin B, and β-actin (all from Santa Cruz Inc., Santa Cruz, CA, USA) at 4°C overnight, followed by incubation with horseradish peroxidase–conjugated secondary antibody (Santa Cruz Inc.) at room temperature for 1 h. The immunoreactive bands were visualized using an enhanced chemiluminescent detection kit (Pierce, Rockford, IL, USA).
Statistical Analysis
Prism V.5.0 software (GraphPad Software, San Diego, CA, USA) was used for statistical analyses. Differences were analyzed by Student’s t-test for two groups or one-way analysis of variance for multiple groups and considered as statistically significant when P values were less than 0.05. The results are expressed as mean ± standard deviation.
Results
Hypoxia Treatment Leaded to Decreased Viability and Increased MIR22HG Expression in AC16 Cardiomyocytes
Initially, the effect of hypoxia on the viability of AC16 cardiomyocytes was explored by CCK-8 assay. We found that exposure to hypoxia for 36 and 48 h both resulted in a decreased viability of AC16 cardiomyocytes compared with normoxia group (Fig. 1A). The expression change of MIR22HG in AC16 cardiomyocytes at different hypoxia time points (12, 24, 36, and 48 h) was examined by quantitative real-time polymerase chain reaction (qRT-PCR). As compared with that in non-treated cells, MIR22HG expression was increased progressively and peaked at 36 h after hypoxia treatment in AC16 cardiomyocytes. However, MIR22HG expression was then reduced following exposure to hypoxia for 48 h in AC16 cardiomyocytes (Fig. 1B). To determine whether MIR22HG was involved in hypoxia-induced injury in AC16 cardiomyocytes, we carried out gain-of-function assays using an overexpression plasmid of MIR22HG and loss-of-function experiments using MIR22HG-specific siRNAs in AC16 cardiomyocytes. As shown in Fig. 1C, D, MIR22HG expression was enhanced in hypoxia or normoxia-treated AC16 cardiomyocytes after transfection with MIR22HG relative to Vector control group, while MIR22HG expression was descended in response to transfection with si-MIR22HG-1 or si-MIR22HG-2 in hypoxia or normoxia-treated AC16 cardiomyocytes compared with that in control group. si-MIR22HG-1 (termed si-MIR22HG) with higher knockdown efficiency was selected for the next experiments.
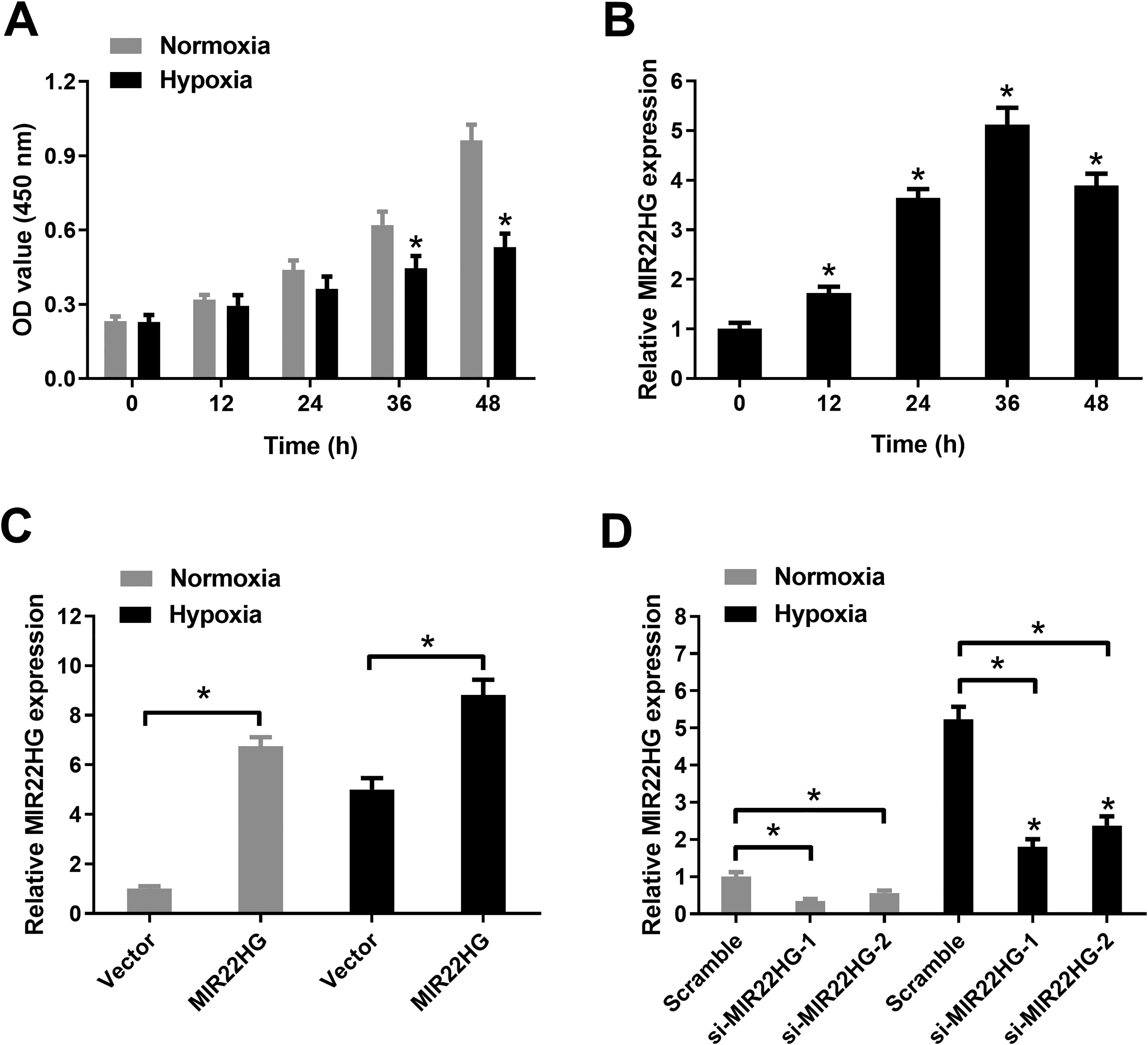
Hypoxia treatment reduced viability and increased MIR22HG expression in AC16 cardiomyocytes. (A) CCK-8 assay was conducted to evaluate cell viability in AC16 cardiomyocytes after treatment with hypoxia or normoxia for 12, 24, 36, and 48 h. (B) MIR22HG expression was detected by qRT-PCR in AC16 cardiomyocytes following exposure to hypoxia for 12, 24, 36, and 48 h. (C) MIR22HG expression was detected by qRT-PCR in AC16 cardiomyocytes after transfection with MIR22HG or Vector, followed by hypoxia or normoxia treatment for 36 h. (D) MIR22HG expression was detected by qRT-PCR after AC16 cardiomyocytes were transfected with si-MIR22HG-1, si-MIR22HG-2, or Scramble prior to hypoxia or normoxia treatment for 36 h. *P < 0.05, n = 3. CCK-8: Cell Counting Kit-8; MIR22HG: miR-22 host gene; qRT-PCR: quantitative real-time polymerase chain reaction.
MIR22HG Overexpression Aggravated Hypoxia-induced Injury in AC16 Cardiomyocytes
The effects of MIR22HG overexpression on hypoxia-induced injury in AC16 cardiomyocytes were explored. As demonstrated by CCK-8 assay, hypoxia-induced viability reduction in AC16 cardiomyocytes was intensified after MIR22HG reconstitution (Fig. 2A). LDH release assay showed that hypoxia challenge led to an increase of LDH release in AC16 cardiomyocytes, which was further aggravated after transfection with MIR22HG (Fig. 2B). The levels of myocardial-specific markers AST and CK-MB were measured to assess myocardial injury. Hypoxia exposure increased the levels of AST (Fig. 2C) and CK-MB (Fig. 2D) in AC16 cardiomyocytes versus that in the normoxia group. However, ectopic expression of MIR22HG further enhanced hypoxia-induced increase of AST (Fig. 2C) and CK-MB (Fig. 2D) levels in AC16 cardiomyocytes. Furthermore, flow cytometry analysis proved that increased expression of MIR22HG reinforced hypoxia-induced apoptosis in AC16 cardiomyocytes (Fig. 2E). MIR22HG re-expression had no effect of the viability, LDH release, levels of AST and CK-MB, and apoptosis of AC16 cardiomyocytes under normoxic condition (Fig. 6A–E). Together, these data suggested that MIR22HG overexpression aggravated hypoxia-induced injury in AC16 cardiomyocytes.
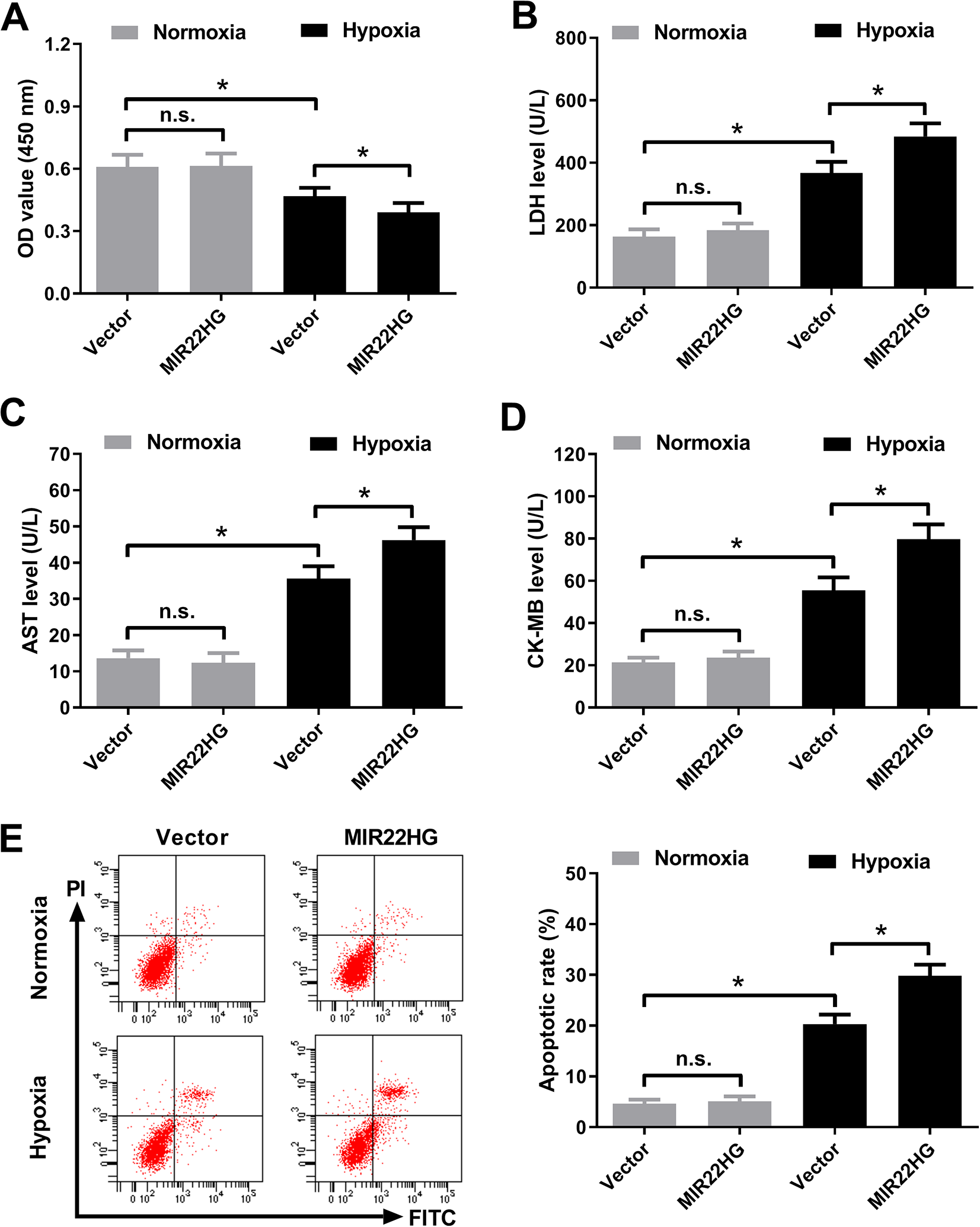
Effects of MIR22HG overexpression on hypoxia-induced injury in AC16 cardiomyocytes. AC16 cardiomyocytes were transfected with MIR22HG or Vector, followed by treatment with hypoxia or normoxia for 36 h. (A) CCK-8 assay was performed to evaluate cell viability in treated AC16 cardiomyocytes. (B) LDH release assay was conducted to measure LDH level in the treated AC16 cardiomyocytes. (C and D) The levels of AST and CK-MB in the treated AC16 cardiomyocytes were detected using respective commercial ELISA kits. (E) The apoptosis of treated AC16 cardiomyocytes was detected using Annexin V-FITC and PI double staining by flow cytometry analysis. *P < 0.05, n = 3. AST: aspartate aminotransferase; CCK-8: Cell Counting Kit-8; CK-MB: creatine kinase-MB; ELISA: enzyme-linked immune sorbent assay; FITC: fluorescein isothiocyanate; LDH: lactate dehydrogenase; MIR22HG: miR-22 host gene; PI: propidium iodide.
MIR22HG Knockdown Attenuated Hypoxia-induced Injury in AC16 Cardiomyocytes
The effect of MIR22HG knockdown on hypoxia-induced injury in AC16 cardiomyocytes was determined. CCK-8 assay demonstrated that interference with MIR22HG abolished the inhibitory effect of hypoxia on the viability of AC16 cardiomyocytes (Fig. 3A). LDH release assay revealed that hypoxia-induced LDH release in AC16 cardiomyocytes was attenuated following silencing of MIR22HG (Fig. 3B). Hypoxia-induced increase of AST (Fig. 3C) and CK-MB (Fig. 3D) levels was weakened in MIR22HG-silencing AC16 cardiomyocytes. Moreover, flow cytometry analysis proved that the percentage of apoptotic AC16 cardiomyocytes was lower in hypoxia + si-MIR22HG group than that in hypoxia + Scramble group (Fig. 3E). MIR22HG depletion did not produce any significant changes on the viability, LDH release, levels of AST and CK-MB, and apoptosis in normoxia-treated AC16 cardiomyocytes. These results collectively suggested that MIR22HG knockdown attenuated hypoxia-induced injury in AC16 cardiomyocytes.
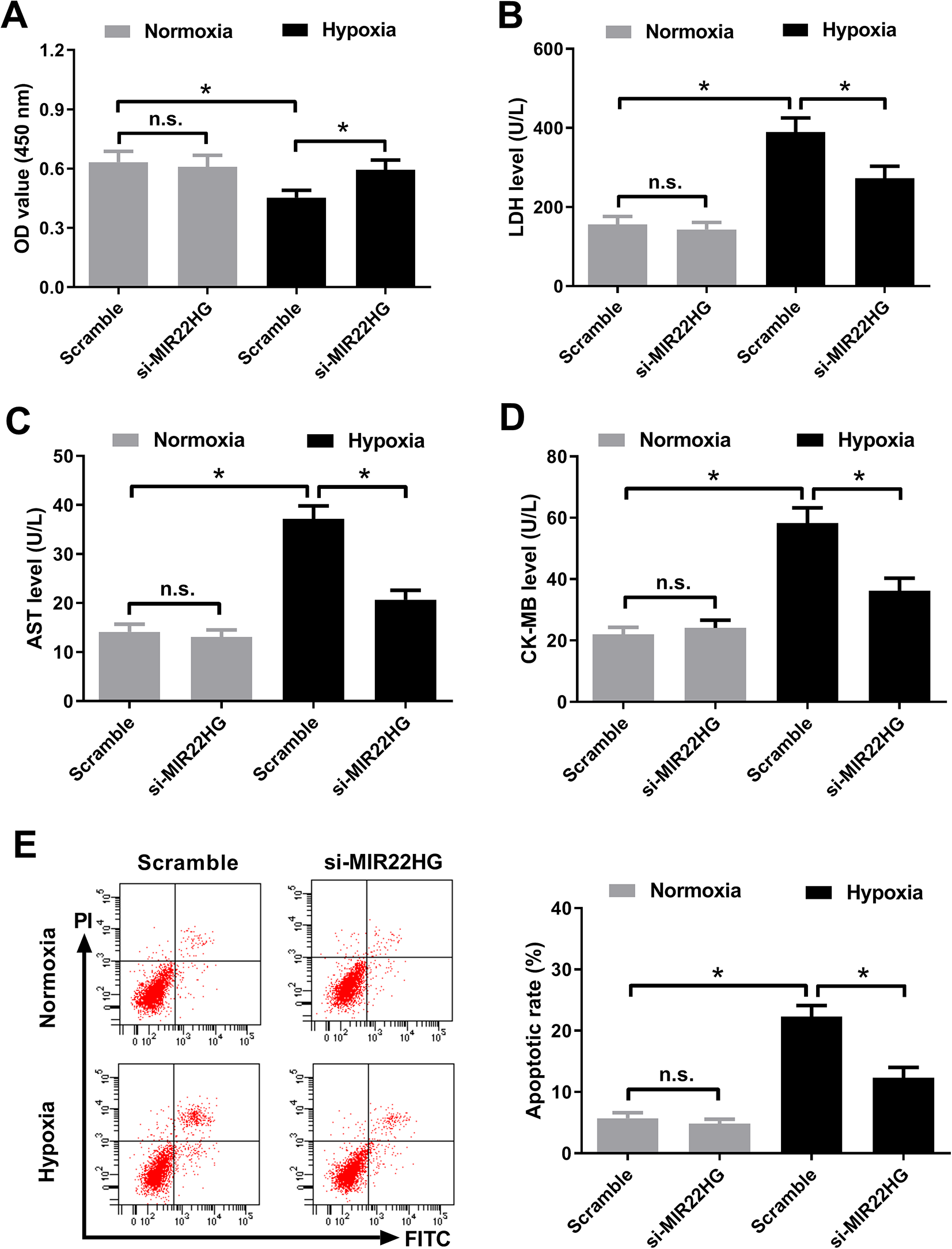
MIR22HG knockdown attenuated hypoxia-induced injury in AC16 cardiomyocytes. AC16 cardiomyocytes were transfected with si-MIR22HG or Scramble prior to challenge with hypoxia or normoxia for 36 h. (A) Cell viability of treated AC16 cardiomyocytes was assessed by CCK-8 assay. (B) LDH released from treated AC16 cardiomyocytes was measured by LDH release assay. (C and D) AST and CK-MB levels in the treated AC16 cardiomyocytes were examined by respective commercial ELISA kits. (E) Apoptosis of treated AC16 cardiomyocytes was analyzed using Annexin V-FITC and PI double staining by flow cytometry analysis. *P < 0.05, n = 3. AST: aspartate aminotransferase; CCK-8: Cell Counting Kit-8; CK-MB: creatine kinase-MB; ELISA: enzyme-linked immune sorbent assay; FITC: fluorescein isothiocyanate; LDH: lactate dehydrogenase; MIR22HG: miR-22 host gene; PI: propidium iodide.
MIR22HG Overexpression Enhanced NF-κB Activation in Hypoxia-treated AC16 Cardiomyocytes
NF-κB pathway has been proposed to be activated after MI 22 and plays a critical role in the development of cardiac remodeling and cardiac dysfunction after MI 23 . Herein, we determined the effect of MIR22HG on the NF-κB pathway in AC16 cardiomyocytes. Western blot analysis of nuclear and cytoplasmic extracts manifested that the nuclear expression of NF-κB p65 and the phosphorylation of IκBα in the cytoplasm were increased, while the cytoplasmic NF-κB p65 expression was decreased in hypoxia-treated AC16 cardiomyocytes (Fig. 4A–D), suggesting the activation of NF-κB pathway in hypoxia-treated AC16 cardiomyocytes. Forced expression of MIR22HG promoted hypoxia-induced increase of levels of nuclear NF-κB p65 and IκBα phosphorylation in AC16 cardiomyocytes. Forced expression of MIR22HG decreased the level of cytoplasmic NF-κB p65 in hypoxia-exposed AC16 cardiomyocytes (Fig. 4A, B). MIR22HG knockdown offset hypoxia-induced elevation of nuclear NF-κB p65 expression and IκBα phosphorylation in AC16 cardiomyocytes, while MIR22HG knockdown increased the level of cytoplasmic NF-κB p65 in hypoxia-exposed AC16 cardiomyocytes (Fig. 4C, D). However, MIR22HG overexpression or knockdown did not affect the levels of cytoplasmic NF-κB p65, nuclear NF-κB p65, and IκBα phosphorylation in AC16 cardiomyocytes under normoxic condition (Fig. 4A–D). Therefore, these results proved that MIR22HG overexpression enhanced NF-κB activation in hypoxia-exposed AC16 cardiomyocytes.
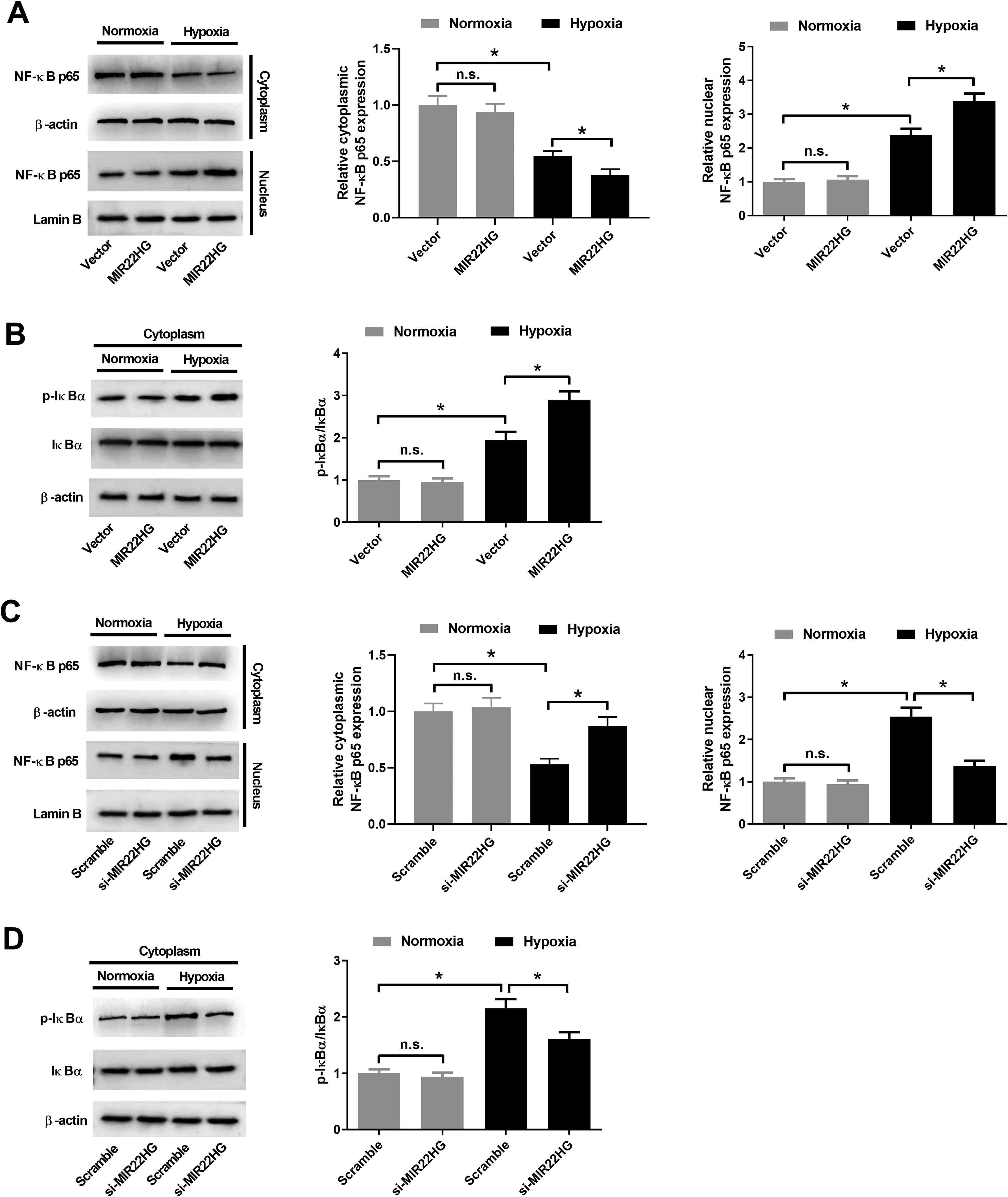
Effect of MIR22HG on the NF-κB pathway in hypoxia-treated AC16 cardiomyocytes. (A and B) AC16 cardiomyocytes were transfected with MIR22HG or Vector prior to hypoxia or normoxia exposure for 36 h, followed by the examination of cytoplasmic NF-κB p65, nuclear NF-κB p65, cytoplasmic p-IκBα, and cytoplasmic IκBα by western blot analysis. (C and D) AC16 cardiomyocytes were transfected with si-MIR22HG or Scramble prior to hypoxia or normoxia exposure for 36 h, followed by the detection of nuclear NF-κB p65, cytoplasmic p-IκBα, and cytoplasmic IκBα by western blot analysis. *P < 0.05, n = 3. MIR22HG: miR-22 host gene; NF-κB: nuclear factor-kappa B.
Inhibition of NF-κB Impaired the Effects of MIR22HG Overexpression on Hypoxia-induced Injury in AC16 Cardiomyocytes
To clarify whether MIR22HG overexpression affected hypoxia-induced injury in AC16 cardiomyocytes via regulating the NF-κB pathway, AC16 cardiomyocytes were transfected with MIR22HG or Vector and then treated with 10 μM PDTC, an inhibitor of NF-κB, and cultured under hypoxic condition for 36 h. The result of CCK-8 assay proved that the promoting effect of MIR22HG overexpression on hypoxia-induced viability reduction in AC16 cardiomyocytes was reversed after the addition of PDTC (Fig. 5A). LDH release assay revealed that MIR22HG overexpression increased LDH release in hypoxia-treated AC16 cardiomyocytes, while such effect was alleviated following administration of PDTC (Fig. 5B). PDTC treatment abrogated the increase of AST (Fig. 5C) and CK-MB (Fig. 5D) levels induced by MIR22HG overexpression in hypoxia-exposed AC16 cardiomyocytes. MIR22HG overexpression-induced apoptosis in AC16 cardiomyocytes under hypoxia condition was impeded due to PDTC treatment (Fig. 5E). In a word, these data suggested that inhibition of NF-κB counteracted the promoting effects of MIR22HG overexpression on hypoxia-induced injury in AC16 cardiomyocytes.
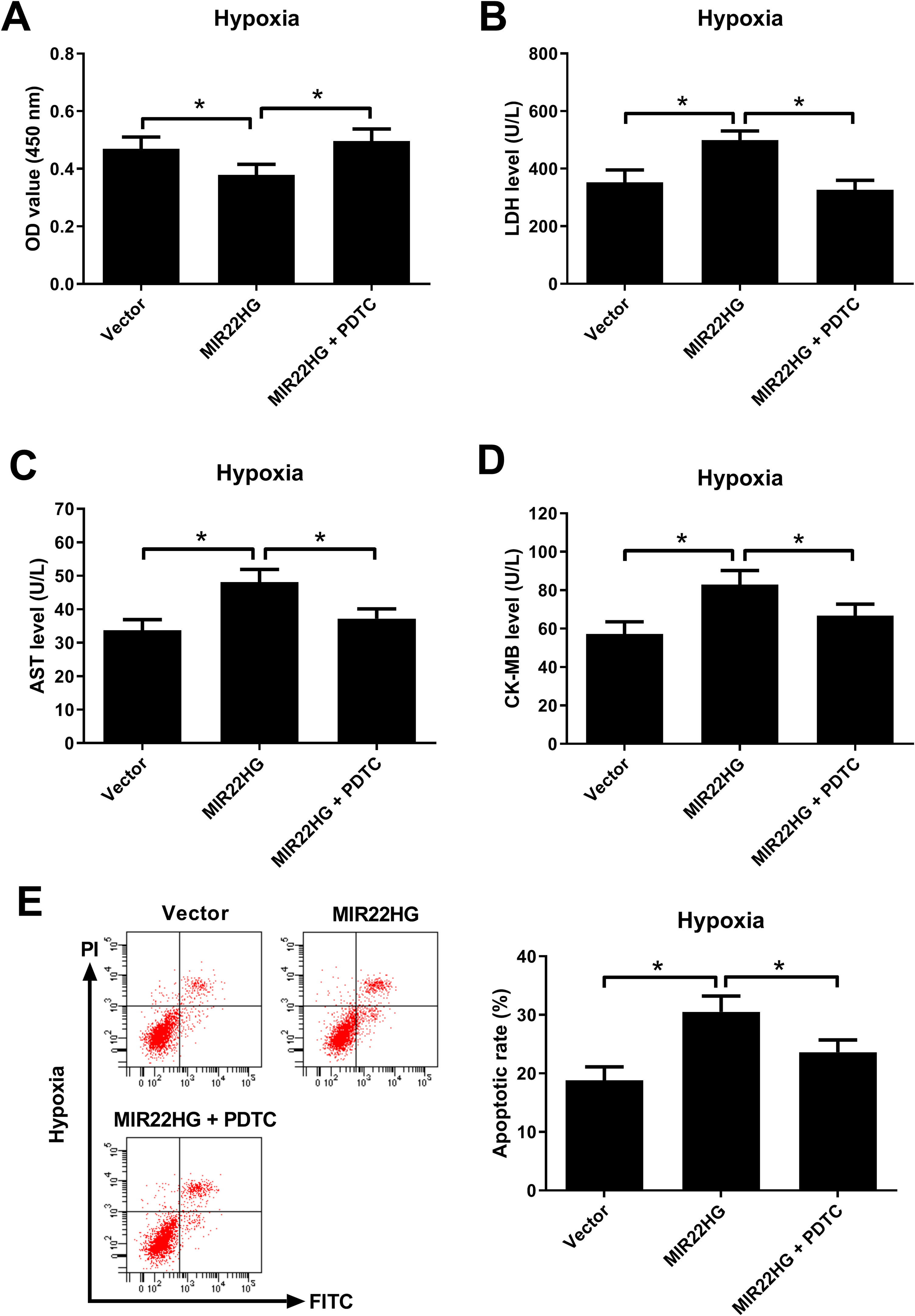
Inhibition of NF-κB antagonized the promoting effects of MIR22HG overexpression on hypoxia-induced injury in AC16 cardiomyocytes. AC16 cardiomyocytes were transfected with MIR22HG or Vector and then treated with 10 μM PDTC, followed by hypoxia treatment for 36 h. (A) Evaluation of cell viability in treated AC16 cardiomyocytes was carried out by CCK-8 assay. (B) LDH released from treated AC16 cardiomyocytes was measured by LDH release assay. (C and D) AST and CK-MB levels in treated AC16 cardiomyocytes were measured using respective commercial ELISA kits. (E) Flow cytometry analysis was performed to determine the apoptosis of treated AC16 cardiomyocytes. *P < 0.05, n = 3. AST: aspartate aminotransferase; CCK-8: Cell Counting Kit-8; CK-MB: creatine kinase-MB; ELISA: enzyme-linked immune sorbent assay; LDH: lactate dehydrogenase; MIR22HG: miR-22 host gene; NF-κB: nuclear factor-kappa B; PDTC: pyrrolidine dithiocarbamate.
MIR22HG Knockdown Inhibited the NF-κB Pathway by Upregulating miR-24 in AC16 Cardiomyocytes
Previous studies have well documented that miR-24-3p, which exerts cardioprotective effects in myocardial ischemia/reperfusion injury 24,25 , is identified as a direct target of MIR22HG 26 . Thus we hypothesized that MIR22HG regulated hypoxia-induced injury in AC16 cardiomyocytes via NF-κB activation by targeting miR-24. We found that hypoxia exposure led to a reduction of miR-24 expression in AC16 cardiomyocytes (Fig. 6A). Moreover, ectopic expression of MIR22HG inhibited miR-24 expression (Fig. 6B), while interference with MIR22HG increased miR-24 expression in hypoxia-exposed AC16 cardiomyocytes (Fig. 6C). Additionally, it was demonstrated that depletion of MIR22HG-mediated increase of cytoplasmic NF-κB p65 and decrease of nuclear NF-κB p65 expression in AC16 cardiomyocytes under hypoxic condition was relieved following reintroduction with anti-miR-24 (Fig. 6D). MIR22HG knockdown-mediated decrease of IκBα phosphorylation in hypoxia-exposed AC16 cardiomyocytes was relieved after reintroduction with anti-miR-24 (Fig. 6E). Accordingly, we concluded that MIR22HG silencing inhibited the NF-κB pathway by increasing miR-24 expression in AC16 cardiomyocytes.
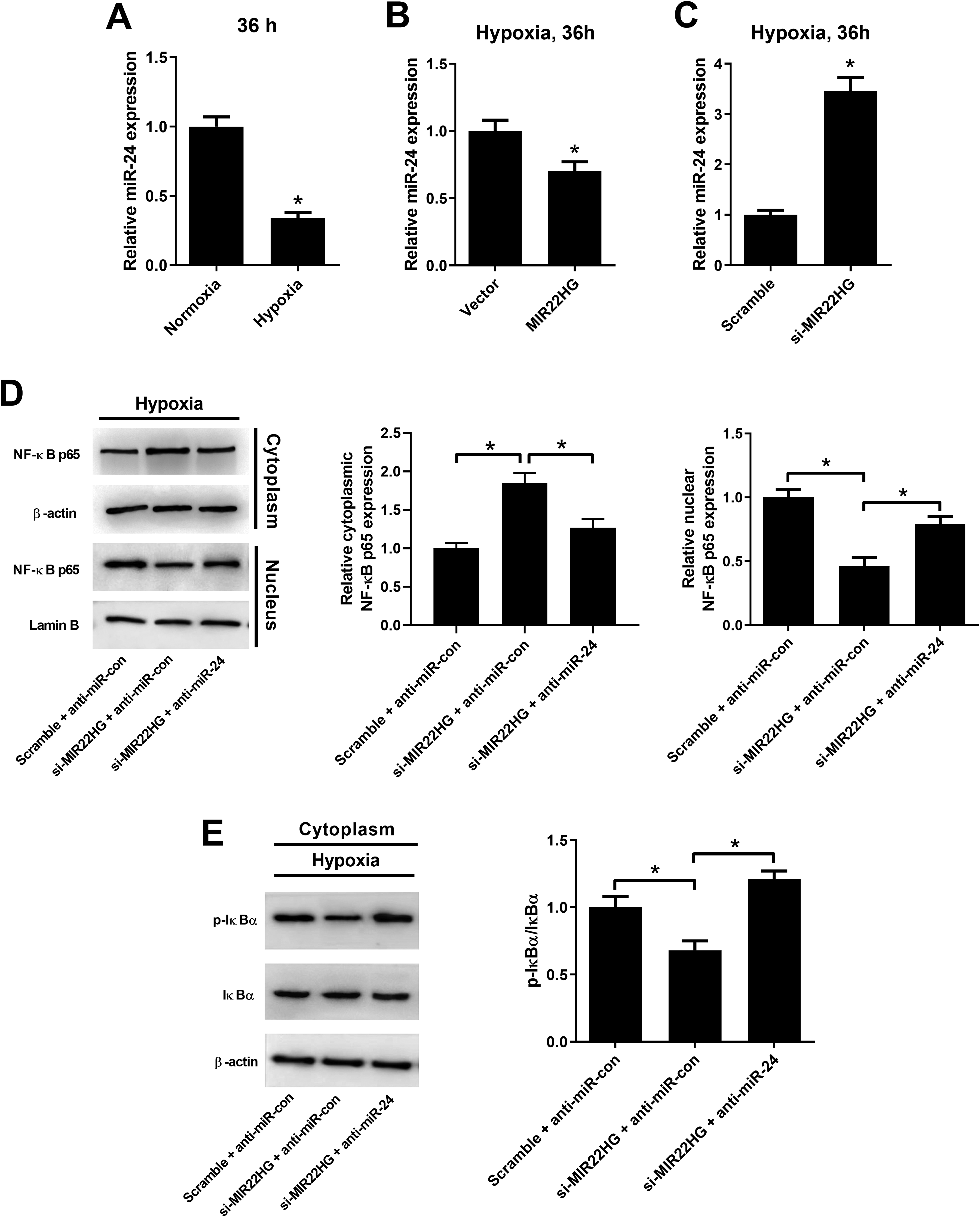
MIR22HG silencing inhibited the NF-κB pathway by increasing miR-24 expression in AC16 cardiomyocytes. (A) qRT-PCR analysis of miR-24 expression in AC16 cardiomyocytes exposed to hypoxia or normoxia for 36 h. (B) qRT-PCR analysis of miR-24 expression in AC16 cardiomyocytes after transfection with MIR22HG or Vector under hypoxic conditions for 36 h. (C) qRT-PCR analysis of miR-24 expression in AC16 cardiomyocytes after transfection with si-MIR22HG or Scramble under hypoxic conditions for 36 h. (D and E) The protein levels of cytoplasmic NF-κB p65, nuclear NF-κB p65, cytoplasmic p-IκBα, and cytoplasmic IκBα were detected by western blot after AC16 cardiomyocytes were transfected with Scramble + anti-miR-con, si-MIR22HG + anti-miR-con, or si-MIR22HG + anti-miR-24 and cultured under hypoxic conditions. *P < 0.05, n = 3. MIR22HG: miR-22 host gene; NF-κB: nuclear factor-kappa B; qRT-PCR: quantitative real-time polymerase chain reaction.
Inhibition of miR-24 Resisted the Effects of MIR22HG Silencing on Hypoxia-induced Injury in AC16 Cardiomyocytes
Next, we determined whether miR-24 was involved in the effects of MIR22HG silencing on hypoxia-induced injury in AC16 cardiomyocytes. The results demonstrated that MIR22HG knockdown promoted the viability (Fig. 7A), decreased the levels of LDH (Fig. 7B), AST (Fig. 7C), and CK-MB (Fig. 7D), and suppressed apoptosis (Fig. 7E) in AC16 cardiomyocytes under hypoxia conditions. These above effects were reversed by miR-24 downregulation. These results demonstrated that inhibition of miR-24 reversed the effects of MIR22HG silencing on hypoxia-induced injury in AC16 cardiomyocytes.
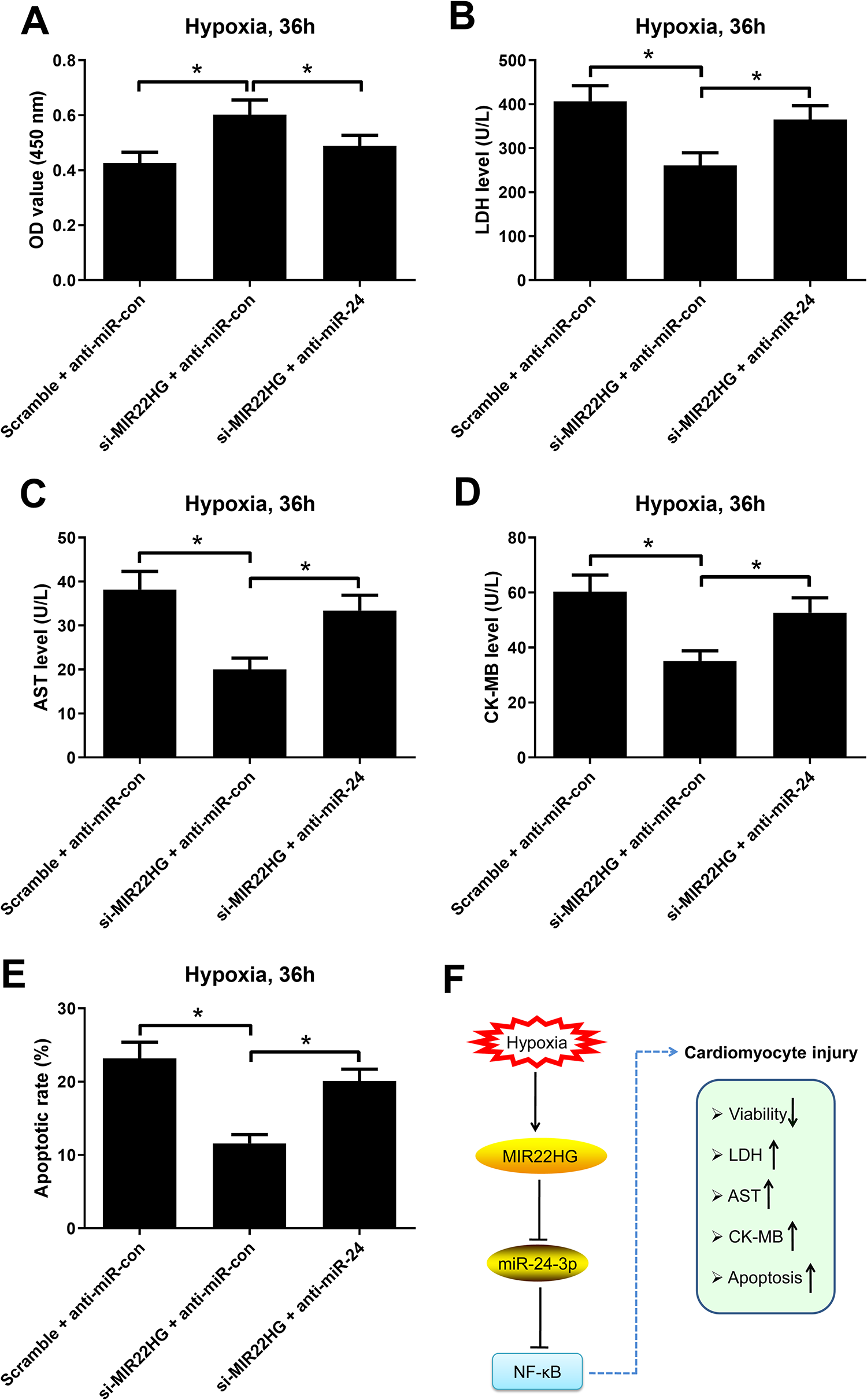
MIR22HG/miR-24 axis regulated hypoxia-induced injury in AC16 cardiomyocytes. AC16 cardiomyocytes were transfected with Scramble + anti-miR-con, si-MIR22HG + anti-miR-con, or si-MIR22HG + anti-miR-24 and cultured under hypoxic conditions, followed by the detection of viability (A), LDH release (B), levels of AST (C) and CK-MB (D), and apoptosis (E) by CCK-8, LDH release assay, respective commercial ELISA kits, and flow cytometry analysis, respectively. *P < 0.05, n = 3. (F) A schematic diagram showing that MIR22HG overexpression aggravated hypoxia-induced injury in AC16 cardiomyocytes via enhancing NF-κB activation by targeting miR-24. AST: aspartate aminotransferase; CCK-8: Cell Counting Kit-8; CK-MB: creatine kinase-MB; ELISA: enzyme-linked immune sorbent assay; LDH: lactate dehydrogenase; MIR22HG: miR-22 host gene; NF-κB: nuclear factor-kappa B.
Discussion
MI is a common presentation for ischemic heart disease caused by an imbalance of oxygen supply and demand, which eventually leads to heart failure and sudden death 27 . Available evidence suggests that MI is largely attributed to hypoxia-induced injury of cardiomyocytes 28 . In spite of the significant improvements in the methods of prevention and treatment for MI, MI is still associated with a substantially higher fatality rate worldwide 29 . Therefore, it is urgently needed to explore preferable effective therapeutic approaches for MI. Multiple lines of evidence has shown that lncRNAs are linked to the development and pathology of various kinds of cardiovascular diseases, including MI 30 . For instance, knockdown of lncRNA AK139328 relieved myocardial ischemia/reperfusion injury in diabetic mice via inhibiting cardiomyocyte autophagy by increasing miR-204-3p expression 31 . Knockdown of Mirt1 attenuated AMI injury presumably by decreasing cardiomyocytes apoptosis and reducing inflammatory cell infiltration through suppressing NF-κB activation 32 . LncRNA TTTY15 attenuated hypoxia-induced cardiomyocyte injury by targeting miR-455-5p 33 . Overexpression of lncRNA HOTAIR alleviated MI or hypoxia-induced cardiomyocytes apoptosis by sponging miR-519d-3p 34 . All these studies collectively suggest the involvement of lncRNAs in the pathogenesis, and progression of MI and lncRNAs can therefore be used as potential therapeutic targets for the treatment of MI.
In the present study, we demonstrated that hypoxia exposure induced injury in AC16 cardiomyocytes by decreasing viability, increasing the leakage of myocardial injury markers LDH, AST, and CK-MB, and eliciting apoptosis in AC16 cardiomyocytes, which was in accordance with the previous studies 35,36 . Hypoxia is recognized as a well-known factor in triggering apoptosis of cardiomyocytes and affects a variety of pathological responses including infarction 37,38 . Our study provided the evidence that hypoxia treatment increased MIR22HG expression in AC16 cardiomyocytes, consistent with the published researches 20,21 . We further demonstrated that MIR22HG re-expression aggravated hypoxia-induced viability reduction, leakage of myocardial injury markers LDH, AST, and CK-MB, and apoptosis in AC16 cardiomyocytes, while MIR22HG knockdown elicited the reverse effects. It is the first time to prove the function of MIR22HG in hypoxia-induced injury in AC16 cardiomyocytes.
NF-κB is a pleiotropic transcription factor consisting of two subunits p65 and p50. Under normal physiological conditions, NF-κB is mainly localized in the cytoplasm as inactive form while bound to its cytoplasmic inhibitory protein (IκBα) 39 . NF-κB is activated in response to external stimuli, leading to the phosphorylation and degradation of IκBα and the subsequent nuclear translocation of NF-κB subunits, where it can stimulate transcription of NF-κB-dependent target genes 39 . The NF-κB pathway has been shown to be implicated in cardiac pathological processes including cardiomyocyte apoptosis 40 . NF-κB also participates in the regulation of the expression of inflammatory mediators and is therefore closely associated with inflammatory response in myocardial ischemia-reperfusion injury 41 . It was reported that NF-κB signaling was activated following myocardial ischemia-reperfusion 42 and inhibition of NF-κB signaling alleviated acute ischemic injury after MI 43,44 . Consistently, our study demonstrated that hypoxia challenge increased nuclear NF-κB p65 expression and IκBα phosphorylation in AC16 cardiomyocytes, suggesting that NF-κB was activated in hypoxia-treated AC16 cardiomyocytes. MIR22HG overexpression further enhanced NF-κB activation in hypoxia-treated AC16 cardiomyocytes. Rescue experiments demonstrated that inhibition of NF-κB pathway by PDTC impaired the effects of MIR22HG overexpression on hypoxia-induced injury in AC16 cardiomyocytes. Thus, we inferred that MIR22HG overexpression aggravated hypoxia-induced injury in AC16 cardiomyocytes by enhancing NF-κB activation.
miR-24, expressed in both cardiomyocytes and fibroblasts, has been recently reported to be associated with post-MI response 45 . However, there are two contrasting results referring to the expressional and function of miR-24 in the mouse acute MI setting. Fiedler et al. reported that miR-24 was enriched in cardiac endothelial cells and upregulated after cardiac ischemia 46 . Blocking of endothelial miR-24 limited myocardial infarct size of mice via preventing endothelial apoptosis and enhancing vascularity 46 . Wei and workmates demonstrated that downregulated miR-24-3p could enhance cell viability, reduce LDH release, and attenuate cell apoptosis in ischemia/reperfusion (I/R)-injured cardiomyocytes 47 . In contrast, Qian et al. showed that miR-24 was an antiapoptotic miRNA that was downregulated in the ischemic zones of the murine LV after MI 48 . miR-24 overexpression inhibited cardiomyocyte apoptosis, attenuated infarct size, and reduced cardiac dysfunction in a mouse MI model 48 . Pan et al. showed that miR-24 expression was decreased in myocardium after AMI and miR-24 agomir reduced cardiomyocyte apoptosis, narrowed infarction area, and improved cardiac function in rats 49 . Our present study proved that miR-24 expression was decreased in response to hypoxia treatment in AC16 cardiomyocytes. Studies have established that lncRNAs can influence the process of MI through functioning as a molecular sponge of miRNAs 50 . For example, MIAT could absorb miR-24 through its sponge-like action as a competing endogenous RNA to govern cardiac fibrosis in MI 51 . A previous study suggested that miR-24 was a target of MIR22HG 26 . Thus, we hypothesized that MIR22HG overexpression aggravated hypoxia-induced injury in AC16 cardiomyocytes by targeting miR-24. We discovered that MIR22HG negatively regulated miR-24 expression in hypoxia-treated AC16 cardiomyocytes. Notably, it was reported that miR-24 inhibited activation of NF-κB in high glucose-induced vascular smooth muscle cells by targeting high mobility group box-1 (HMGB1) 52 . HMGB1 is known to interact with cell-surface receptors, resulting in activation of NF-κB pathway 53 . A previous study showed that miR-24 targeted multiple components of pathogen recognition receptor signaling to inhibit NF-κB activation 54 . Our study demonstrated that miR-24 downregulation abolished the inhibitory effect of MIR22HG knockdown on the NF-κB pathway in AC16 cardiomyocytes, suggesting that MIR22HG knockdown inhibited the NF-κB pathway in AC16 cardiomyocytes by targeting miR-24. In addition, it was manifested that inhibition of miR-24 reversed the effects of MIR22HG silencing on hypoxia-induced injury in AC16 cardiomyocytes. Therefore, we concluded that MIR22HG silencing attenuated hypoxia-induced injury in AC16 cardiomyocytes via inactivation of the NF-κB pathway by upregulating miR-24.
In summary, our study demonstrated that hypoxia exposure increased MIR22HG expression in AC16 cardiomyocytes. Moreover, we provided the first evidence that MIR22HG overexpression aggravated hypoxia-induced injury in AC16 cardiomyocytes via enhancing NF-κB activation by targeting miR-24. Fig. 7F describes a possible function and mechanism of MIR22HG in hypoxia-induced cardiomyocyte injury, providing new insights into the pathogenesis of MI. The discovery of this mechanism may shed light on the clinical diagnosis and therapy for MI.
Footnotes
Authors' Contributions
XY and JH collected key background information, designed and performed the experiments, analyzed the experimental results, and drafted the manuscript.
Statement of Human and Animal Rights
This article does not contain any studies with human or animal subjects.
Statement of Informed Consent
There are no human subjects in this article and informed consent is not applicable.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.