
Abstract
Treatment of high-risk paediatric neuroblastoma represents an unmet clinical need. Chimeric antigen receptor-modified T cell (CAR-T) therapy is a promising treatment option, but there exist some challenges regarding specificity and potency. The current study used ganglioside GD2 as a target for CAR-T construction because of its selective overexpression in neuroblastoma cells. We engineered a GD2-based CAR-T construct, including ICOS and 4-1BB co-stimulatory domains for better persistence. The cytotoxicity of the generated CAR-T cells (PG3-GD2-CAR-T) was verified using in vitro and in vivo assays. PG3-GD2-CAR-T cells exerted potent anti-tumour activity in vitro and in vivo, with minimal effects on peripheral blood cells. PG3-GD2-CAR-T cells exhibited encouraging specificity for and potency against neuroblastoma.
Keywords
Introduction
Neuroblastoma is one of the most common extra-cranial solid tumours in children, with 90% of patients diagnosed during the first 10 years of life, with a median age at diagnosis of 18 months. 1 It is a neuroendocrine tumour that occurs during the development of the sympathetic nerves. 2 The clinical manifestations of neuroblastoma are extremely variable. In young infants, many tumours spontaneously regress without the need for treatment; in contrast, patients older than 18 months of age at diagnosis frequently present with wide-spread metastatic disease. 3 Although the survival rates for patients with low- and medium-risk neuroblastoma have improved, 4 for patients with high-risk tumours (patient older than 18 months, with the degree of risk determined according to the tumour grade, degree of metastatic disease, and genetic factors), the treatment options remain poor.5 –8 The most effective strategy for these patients, at present, is to introduce monoclonal antibodies (MAbs) against ganglioside GD2 (anti-GD2 MAbs) as an immunotherapy treatment.7,9 –14
GD2 is highly expressed in a variety of paediatric and adult tumours, including neuroblastoma, glioma and melanoma.15,16 GD2 is usually expressed during foetal development, and its expression in normal post-natal tissues is low, usually limited to osteoprogenitors, the brain, peripheral nerves, and skin melanocytes. 17 Based on these characteristics of GD2, a number of GD2-specific immunotherapy strategies have been developed, including specific antibodies, drug coupling and chimeric antigen receptor-modified T cell therapy.13,14,18 –21
Although the introduction of anti-GD2 monoclonal antibodies can temporarily improve the survival of neuroblastoma patients, approximately 50% of them will eventually relapse and die. 7 These patients need more effective and targeted therapies. One alternative strategy is chimeric antigen receptor-modified T cell (CAR-T) therapy. Recently, the use of CAR-T therapy has demonstrated exciting results against haematological malignancies, 22 and it has also shown promise in the treatment of neuroblastoma. CARs commonly contain three modules: an extracellular antigen-binding module, a transmembrane domain (TM domain), and an intracellular signalling structure that transmits activation signals. The TM domain anchors the CAR structure in the cell membrane and is commonly derived from T cell function-regulating molecules such as CD8 and CD28. The intracellular signalling structure typically consists of the T cell receptor (TCR) CD3ζ signalling chain connected to one or two co-stimulation signals, such as CD28, 4-1BB, OX40 and ICOS.
Recently, Prapa et al. constructed a second-generation CAR structure with a mouse-derived GD2 single chain variable fragment (scFv) linked with the 4-1BB ligand as its co-stimulatory domain and examined the anti-tumour activity of this GD2-CAR-T against neuroblastoma. 21 While the authors verified the feasibility of GD2 as a target for neuroblastoma, the CAR-T cells constructed with their CAR structure have been shown to exhibit limited cytotoxicity, with poor in vitro efficacy because of inefficient target ratios. Richman et al. tested three modifications to the scFv fragment. First, they linked the scFv fragment of a mouse-derived GD2 antibody, 14G2a, to 4-1BB; second, they engineered a mutation into the VH domain to increase the affinity of the scFv fragment; third, they increased the length of the linker between VL and VH to improve scFv fragment stability. Based on these modifications, they constructed three CAR structures and investigated the effects of different CAR structures on the anti-tumour activity of CAR-T cells. Their research shows that increasing the affinity of scFv fragments can effectively enhance the anti-tumour activity of CAR-T cells, but it is accompanied by very serious “on-target, off-tumour tissue” side effects. However, increasing the length of the linker between VL and VH improved the ability of CAR-T cells to efficiently home in on the tumour site, but there was hardly any improvement on the anti-tumour activity of the CAR-T cells. It has been speculated that the production of CAR-T cells may be exhausted too quickly to continuously inhibit the proliferation of tumour cells. 18 Therefore, it is necessary to design a CAR structure with high cytotoxicity and low side effects that can also continuously and efficiently exert its anti-tumour activity.
In 2018, Guedan et al. reported for the first time that ICOS was used in combination with 4-1BB to construct a third-generation CAR containing two co-stimulatory molecules. They found that compared with CAR structures constructed using CD28 as an anchoring domain, introduction of the intracellular and transmembrane regions of ICOS can promote the differentiation of CD4+ T cells into a Th1 / Th17 phenotype, thereby increasing the anti-tumour activity of CD8+ T cells. At the same time, introduction of 4-1BB molecules as part of the CAR structure could effectively prolong the persistence of CD8+ T cells. 23
Here, we describe the construction of a novel third-generation anti-GD2-CAR vector. We linked the scFv fragment of a humanised GD2 antibody to the transmembrane and intracellular domains of ICOS using human Fc fragments. The intracellular domain of ICOS is the initial co-stimulatory molecule, which is connected to the intracellular region of 4-1BB, as the second co-stimulatory molecule of the CAR structure, and both are linked to CD3ζ. We then tested the efficacy of these structures, in the form of PG3-GD2-CAR-T cells, against neuroblastoma cell lines and mouse xenograft models.
Materials and methods
Cell lines and cell cultivation
The human neuroblastoma cell lines SK-N-SH, SK-N-AS and SH-SY5Y were obtained from PersonGen Biotherapeutics Co., Ltd. SK-N-SH cells were cultured in MEM medium (Thermo Fisher Scientific), supplemented with 10% foetal bovine serum (FBS; Gibco), 1% non-essential amino acids (NEAA; Gibco), and 1 mM sodium pyruvate (NaP; Gibco). SK-N-AS cells were cultured in DMEM (Thermo Fisher Scientific), supplemented with 10% FBS and 1% NEAA. SH-SY5Y cells were cultured in MEM / F-12 (1: 1) (Thermo Fisher Scientific), supplemented with 10% FBS, 1% NEAA, and 1 mM NaP. Jurkat cells (American Type Culture Collection, ATCC) were cultured in RPMI-1640 medium (Thermo Fisher Scientific), supplemented with 10% FBS. T cells extracted from the peripheral blood of healthy donors were cultured in TexMacsTM GMP medium (Miltenyi Biotec), supplemented with 155 U/ml IL-7 and 190 U/ml IL-15 (Novoprotein). HEK293 cells (ATCC) were cultured in DMEM supplemented with 10% FBS. All cells were cultured at 37°C in a humidified atmosphere containing 5% CO2.
PG3-GD2-CAR construction
In our study, we constructed a novel third-generation anti-GD2-CAR vector, which contained an scFv fragment of a humanised GD2 antibody, a human Fc fragment, a human ICOS transmembrane region and intracellular region, a human 4-1BB intracellular domain, and human CD3ζ sequence.The GD2-CAR cassettes was later sub-cloned into the pHULK PiggyBac Mammalian Expression Vector and designated as the PG3-GD2-CAR plasmid.
Lentivirus transduction
Lentivirus preparation has been well documented in our previous work. 24 CAR-T cells were constructed as follows: healthy human donor peripheral blood was obtained and peripheral blood mononuclear cells (PBMCs) were isolated using the Ficoll method. Briefly, blood was diluted 1:1 with phosphate buffer solution (PBS), and then added to the Ficoll (diluted blood:Ficoll = 2:1). The sample was then centrifuged (BECKMAN COULTER, Inc., CA, USA, Allegra X-12R centrifuge) at 800 × g for 30 min, and the PBMC layer was removed. The monocyte layer was drawn and placed in a 24-well plate for propagation with approximately 5 × 106 T cells, 1.5 ml of culture medium, and 20 μl anti-CD3/CD28 beads (Miltenyi Biotec) per well. After a 48-h activation, 5 × 105 T cells were transferred to a 48-well plate and transduced with concentrated virus supernatant (multiplicity of infection (MOI) = 10), in a final well volume of 200 μl. After 16 h, the supernatant was removed, and culture medium was added. The resulting transfected cells were referred to as PG3-GD2-CAR-T cells. T cells were propagated throughout this process in TexMacsTM GMP medium supplemented with IL-7 and IL-15.
Construction of PG3-GD2-CAR-Jurkat cells
We constructed PG3-GD2-CAR-Jurkat cells (PG3-GD2-CAR overexpressed in Jurkat cells) to demonstrate the specificity of the PG3-GD2-CAR vector. Jurkat cells were plated in a 48-well plate at a density of approximately 1 × 106 cells per well and transduced with concentrated PG3-GD2-CAR virus supernatant. After 14 h of transduction, the supernatant was removed and culture medium was added. Subsequently, cells were incubated with APC-conjugated anti-human IgG Fc antibodies (Jackson ImmunoResearch) and sorted with a FACS Aria™III Cell Sorter (BD Biosciences) until the positive rate ⩾ 90%.
Flow cytometry and western blotting
To detect GD2-CAR expression on the surface of T cells and Jurkat cells, we stained PG3-GD2-CAR-T cells and PG3-GD2-CAR-Jurkat cells with APC-conjugated anti-human IgG Fc antibodies. To detect GD2 antigen expression on the surface of neuroblastoma cells, we stained SK-N-SH, SK-N-AS and SH-SY5Y cells with PE-conjugated GD2 antibodies (BD Biosciences). Specifically, the cells were incubated with the antibodies at 37°C for 20 min, washed twice with PBS, and then analysed using a flow cytometer (BD Biosciences). PG3-GD2-CAR-T cells were used for total protein extraction, followed by western blot analysis using mouse-derived anti-CD3ζ antibody (BD Biosciences) as the primary antibody and goat anti-mouse horseradish peroxidase (HRP) antibody (Solarbio, Beijing, China) as the secondary antibody. This western blotting method was performed as previously described. 25
In vitro cytotoxicity and detection of cytokines
We used the xCELLigence real-time cell analyser (RTCA, ACEA) to detect the anti-tumour activity of PG3-GD2-CAR-T cells against neuroblastoma cells in vitro. For this assay, 1 × 104 target cells (SK-N-SH, SK-N-AS, or SH-SY5Y) were plated (ACEA,catalog #00300600890) in a culture volume of 100 μl/well and cultured for 24 h. Then the effector cells (PG3-GD2-CAR-T) were added at effector:target (E:T) ratios of 1:2, 1:1 and 4:1, and co-cultured for 48 h at a final well volume of 200 μl. The cell index was monitored every 15 min during this time period. After incubation for 48 h, the supernatant was aspirated to detect the specific cytokine profiles using a cytometric bead array (CBA) system (BD Biosciences).
Xenotransplantion models
To assess in vivo anti-tumour activity of PG3-GD2-CAR-T cells, we generated a xenograft mouse model using the NOD-PrkdcscidIl2rgtm1/Bcgen (B-NSG) mouse (Biocytogen) injected with human SK-N-AS neuroblastoma cells. Six- to eight-week-old female mice, five per group, were injected subcutaneously on day 0 with 1.5 × 106 SK-N-AS cells suspended in 0.1 ml Matrigel (BD Biosciences), and on day 7, either T cells only or PG3-GD2-CAR-T cells (1.5 × 106 cells per mouse, CAR positive rate of 35%) were injected into the tail vein. Animal weight and tumour growth were monitored up to the end of the in vivo study or the point at which tumours reached 1000 mm3. Tumour lengths and widths were recorded, and the tumour volume was calculated according to the following formula (volume = (length × width 2 )/2).
Histology and TUNEL staining
The tumour tissues of two mice in the T cell group and two mice in the PG3-GD2-CAR-T group were embedded in paraffin and 3-μm sections were Entrusted to Suzhou Cancercell Biotechnology Co., Ltd. and prepared for immunohistochemical analysis. The paraffin sections were stained with haematoxylin and eosin (H&E) to observe pathological changes in tumour tissues; stained with anti-CD3 antibody to observe the infiltration of T cells in tumour tissue; and stained with a TUNEL kit to observe the apoptosis of tumour cells in the tissue.
Data analysis
Statistical analyses were performed using GraphPad Prism Software (version 5.0). Paired t-tests were used to compare differences between groups. The brackets in the figures indicate which groups were compared, and *, ** and *** indicate p < 0.05, < 0.01 and < 0.001, respectively.
Results
Construction and specificity verification of GD2-CAR vector
To construct a novel third-generation anti-GD2-CAR vector, we linked the scFv fragment of a humanised GD2 antibody to the transmembrane and intracellular domains of ICOS using human Fc fragments. The intracellular domain of ICOS is the first co-stimulatory molecule, which is connected to the intracellular region of 4-1BB, as the second co-stimulatory molecule of the CAR structure, and they are then linked to CD3ζ. Figure 1(a) shows a schematic diagram of this CAR structure. The PG3-GD2-CAR structure was later subcloned into a lentiviral expression vector and named the PG3-GD2-CAR plasmid.
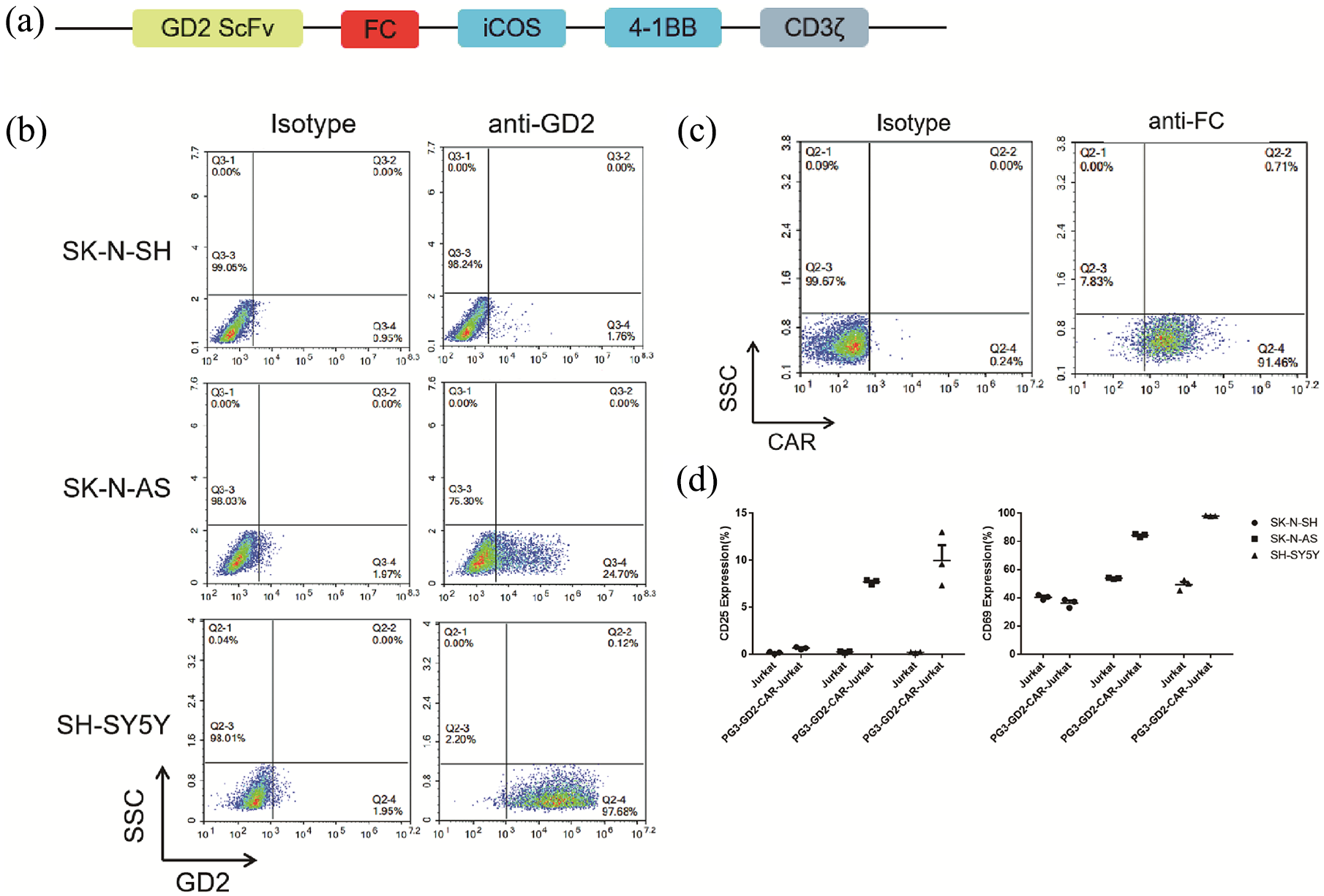
Construction and specificity verification of GD2-CAR vector. (a) Schematic representation of the PG3-GD2-CAR vector. The PG3-GD2-CAR construct contains a scFv fragment of humanised GD2 antibody, a hinge domain (Fc), two co-stimulatory domains (ICOS and 4-1BB), and the intracellular signalling domain CD3ζ. (b) Flow cytometry analysis of cell-surface expression of GD2 on neuroblastoma cell lines. (c) Flow cytometry analysis of cell-surface expression of PG3-GD2-CAR on PG3-GD2-CAR-Jurkat cells. (d) Flow cytometry analysis of cell-surface expression of CD25 and CD69 on Jurkat and PG3-GD2-CAR-Jurkat cells after incubation with neuroblastoma cells at an E:T ratio of 1:1 for 24 h.
We began testing this system by assessing the SK-N-SH, SK-N-AS and SH-SY5Y target cell lines for their GD2 expression, to estimate their response to being challenged by CAR-T cell activity (Figure 1(b)). High GD2 expression was observed on SH-SY5Y cells, while low levels were detected on SK-N-AS cells. The SK-N-SH cell line showed undetectable GD2 levels and was used as negative control.
Jurkat cells were collected and transduced with concentrated PG3-GD2-CAR virus supernatant to construct PG3-GD2-CAR-Jurkat cells. Subsequently, cells were incubated with APC-conjugated anti-human IgG Fc antibodies and sorted until the positive rate ⩾ 90%. The results showed that more than 90% of the cells were CAR+ cells (Figure 1(c)). Jurkat cells and PG3-GD2-CAR-Jurkat cells were then co-incubated with SK-N-SH (GD2neg), SK-N-AS (GD2low), or SH-SY5Y (GD2high) cells at an E:T ratio of 1:1 for 24 h to detect the expression of CD25 and CD69 on the surface of the Jurkat and PG3-GD2-CAR-Jurkat cells. The expression of CD25 and CD69 on the surface of PG3-GD2-CAR-Jurkat cells was significantly increased after co-incubation with SK-N-AS (GD2low) and SH-SY5Y (GD2high) cells when compared with Jurkat cells. In contrast, the expression of CD25 and CD69 on the surface of CAR-Jurkat cells exhibited almost no change when co-incubated with SK-N-SH (GD2neg) cells (Figure 1(d)). These results showed that the PG3-GD2-CAR-Jurkat cells that we constructed can be specifically activated by GD2-positive but not GD2-negative tumour cells.
Construction of PG3-GD2-CAR-T cells
PBMCs from healthy donors were activated for 48 h and transduced with concentrated PG3-GD2-CAR virus supernatant to construct PG3-GD2-CAR-T cells. After 6 to 10 days of culture, the expression of CAR protein on the surface of the T cells was detected by anti-Fc antibodies, and approximately 40% (±10%) of T cells were shown to be CAR+ (Figure 2(a)). To confirm CAR expression in the T cells, western blotting was performed to examine CD3ζ fusion protein expression. Internalised CD3ζ has a molecular weight of approximately 15 kDa, with the dimer being 30 kDa, and the expression of exogenous fusion proteins was clearly identified at 75 kDa in PG3-GD2-CAR-T cells (Figure 2(b)).

Construction of PG3-GD2-CAR-T cells. (a) Flow cytometry analysis of cell-surface expression of PG3-GD2-CAR on PG3-GD2-CAR-T cells. (b) Western blot analysis of PG3-GD2-CAR expression in PG3-GD2-CAR-T cells. From left to right: the corresponding position of the protein in PG3-GD2-CAR-T and T cells. As shown, the exogenous CD3ζ fusion proteins of PG3-GD2-CAR-T cells were in the molecular weight range of 70 to 100 kDa.
PG3-GD2-CAR-T cells exert specific in vitro cytotoxicity against neuroblastoma cells
We used the xCELLigence RTCA (Eisenbio) to detect the anti-tumour activity of PG3-GD2-CAR-T cells against neuroblastoma cells in vitro. Effector (T and PG3-GD2-CAR-T) and Target (SK-N-SH, SK-N-AS, and SH-SY5Y) cells were co-incubated at E:T ratios of 1:2, 1:1 and 4:1 for 48 h. PG3-GD2-CAR-T cells significantly inhibited the growth of GD2+neuroblastoma cells, and the inhibitory effect on SH-SY5Y cells (which have high GD2 expression) is particularly obvious (Figure 3(a) second and third rows), when compared with T cells. For GD2- neuroblastoma cells, there was no significant difference between the T cell group and the PG3-GD2-CAR-T group (Figure 3(a) first row). We subsequently analysed the secretion of selected cytokines in the supernatant after co-incubation. The results showed that the secretion of certain cytokines was significantly increased in the supernatant of the PG3-GD2-CAR-T group, including Granzyme-B, TNF-α, IFN-γ, and IL-2 (Figure 3(b)).

PG3-GD2-CAR-T cells exert specific in vitro cytotoxicity against neuroblastoma cells. (a) An xCELLigence real-time cell analyser (RTCA) was used to detect the anti-tumour activity of PG3-GD2-CAR-T cells against neuroblastoma in vitro. Effector (PG3-GD2-CAR-T) and target (SK-N-SH, SK-N-AS or SH-SY5Y) cells were co-cultured at E:T ratios of 1:2, 1:1 and 4:1 for 48 h. The cell index was monitored every 15 min. (b) Cytokine secretion in the supernatant after co-incubation was assessed using a cytometric bead array (CBA) system.
GD2 CAR-T cells provide potent therapeutic activity in vivo
In the next set of experiments, we generated a mouse xenograft model using the human SK-N-AS neuroblastoma cell line as a target to assess the in vivo anti-tumour activity of PG3-GD2-CAR-T cells. The experimental scheme of is shown in Figure 4(a). SK-N-AS cells were injected subcutaneously into B-NSG mice that were then injected 1 week later with either PG3-GD2-CAR-T cells or control T cells, via the tail vein. The results showed that in mice treated with T cells, tumour development was observed in all mice. In contrast, treatment with PG3-GD2-CAR-T cells significantly suppressed tumour growth in all mice, and in particular, the tumours of three mice completely disappeared (Figure 4(b) and (d)). This indicates that PG3-GD2-CAR-T cells can successfully home in on tumour tissues and exert a continuous anti-tumour effect in vivo. At the same time, there were no significant differences in the counts of red blood cells, white blood cells, and platelets, or in haemoglobin levels, between the PG3-GD2-CAR-T cell group and the T cell group (Figure 4(c)). This indicates that PG3-GD2-CAR-T cells have minimal effect on the blood phase in the peripheral blood of mice, which suggests that they are relatively safe.

GD2 CAR-T cells provide potent therapeutic activity in vivo. (a) Schematic representation of a neuroblastoma xenograft model. (b) Tumor volume growth and weight of tumour to body ratio at sacrifice. *p = 0.0103, **p = 0.0049 by t-test, mean ± SEM. (c) Detection of red blood cells, white blood cells, haemoglobin, and platelets in peripheral blood at sacrifice. Ns: no significant difference by t-test, mean ± SEM. (d) Comparison of tumour size of mice in CAR-T group and T cell group at sacrifice.
Harvested tumours were then analysed by H&E staining, which revealed changes in the histological architecture of specimens treated with PG3-GD2-CAR-T cells versus controls (Figure 5(a)). Immunohistochemical analyses of CD3 were also performed and clusters of infiltrating CD3 positive cells were observed only in tumour sections taken from PG3-GD2-CAR-T cell-treated animals (Figure 5(b)). Finally, TUNEL staining was applied to assess the apoptosis of tumour cells. Apoptotic cells were sparse in tumour specimens of mice treated with T cells, whereas they were clearly visible in the tumour specimens that were treated with PG3-GD2-CAR-T cells (Figure 5(c)).
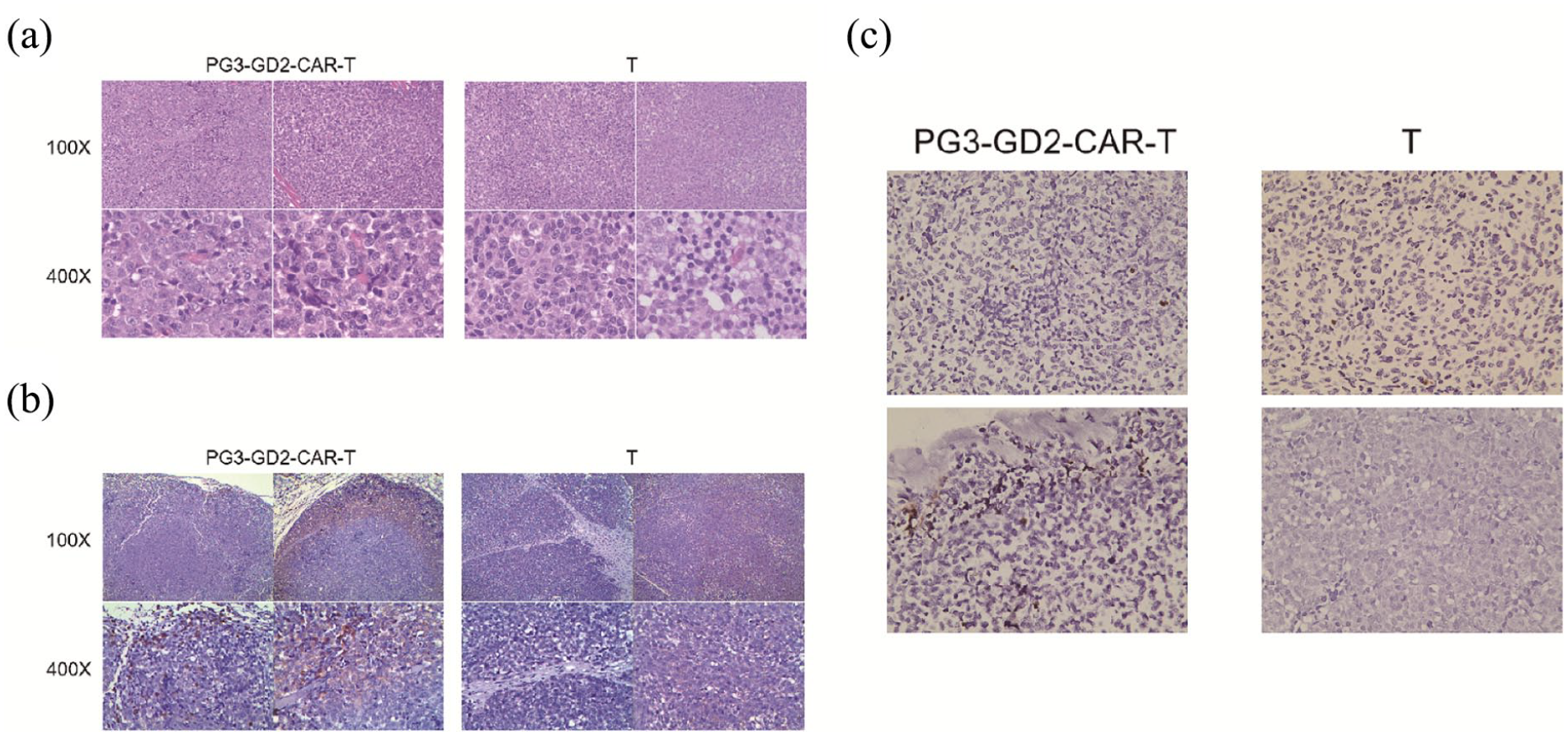
Hematoxylin and eosin staining, immunohistochemistry, and TUNEL assay staining of tumour sections. (a) H&E staining of tumour tissues treated by PG3-GD2-CAR-T cells versus controls. On the left are two tumours from the PG3-GD2-CAR-T group and on the right are two tumours randomly selected from the T cell group. (b) Immunohistochemical analyses of CD3. On the left are two tumours from the PG3-GD2-CAR-T group and on the right are two tumours randomly selected from the T cell group. (c) TUNEL analysis showing apoptotic cells.
Discussion
It is important for the safety and feasibility of CAR-T cell therapy to choose a favourable target. Given its high expression in a variety of paediatric and adult tumours, including neuroblastoma, and its low expression in normal post-natal tissues,15 –17 GD2 has been exploited for a variety of immunotherapeutic strategies including CAR-T therapy.13,14,18,19 –21 The data generated thus far using GD2-CAR-T have demonstrated the feasibility of GD2 as a target for the treatment of neuroblastoma. However, CAR-T is not as effective in solid tumours as it is in haematological malignancies, and there are many challenges in designing CAR-T cells for use against solid tumours. These include a lack of ideal targets, difficulty in T cell homing, and poor persistence and potency of CAR-T cells caused by the immunosuppressive tumour microenvironment. 3 In the current study, we constructed a novel third-generation anti-GD2-CAR vector. We linked the scFv fragment of a humanised GD2 antibody to the transmembrane and intracellular domains of ICOS using human Fc fragments. ICOS and 4-1BB were used as co-stimulatory molecules, linked to CD3ζ. The incorporation of ICOS and 4-1BB as co-stimulatory molecules in a CAR structure can strongly enhance both the persistence and anti-tumour activity of CAR-T cells. 23
Our results demonstrated that PG3-GD2-CAR-T cells showed significant anti-tumour activity against GD2+ neuroblastoma cells both in vivo and in vitro. It was found that PG3-GD2-CAR-T cells exert increased cytotoxicity against GD2-positive cells (SK-N-AS,SH-SY5Y) than active T cells. To explore the associated mechanism, Granzyme-B, TNF-α, IFN-γ and IL-2 were examined; PG3-GD2-CAR-T cells were shown to secrete more Granzyme-B, TNF-α, IFN-γ, and IL-2 when cultured with GD2-positive cells, indicating the target specificity of PG3-GD2-CAR-T cells. Meanwhile, in contrast to that observed for active T cells, PG3-GD2-CAR-T cells could secrete more Granzyme-B, TNF-α, IFN-γ, and IL-2 when killing GD2-negative cells(SK-N-SH), which was possibly due to the fact that modified CARs promoted the activation of T cells (Figure 3(b) first row).
Furthermore, we demonstrated that PG3-GD2-CAR-T cells significantly inhibited disease progression in mouse xenograft models of neuroblastoma cells. The results shows that in comparison with mice treated with T cells, the tumour masses in mice treated with PG3-GD2-CAR-T cells were significantly suppressed, and Histology revealed that there were more infiltrating T cells, structurally damaged tumour tissue, and apoptotic tumour cells observed in the tumour masses. This indicated that PG3-GD2-CAR-T cells could successfully home in on tumour tissues and exert a continuous anti-tumour effect as the tumours of three mice completely disappeared without recurrence. In this in vivo study,we also observed that there was no significant difference in red blood cells, white blood cells, haemoglobin, and platelets between the PG3-GD2-CAR-T cell group and the T cell group, which illustrates that PG3-GD2-CAR-T cells have minimal influence on the blood phase in the peripheral blood of mice, and are relatively safe.
One limitation of our study is that the infiltration of T cells in tumour tissue is limited to the outside rather than the inside. In addition, the number of mice in animal experiments also needs to be increased. Future studies will focus on improving the therapeutic effect of CAR-T on solid tumours, by constructing a fourth-generation GD2-CAR vector that co-expresses PD-1 antibody or IL-15, to further disrupt the tumour microenvironment and improve CAR-T activity and prolong the persistence of CAR-T cells in vivo.
Conclusion
We constructed novel third-generation anti-GD2-CAR-T cells (containing ICOS and 41BB co-stimulatory signals) based on the GD2 humanised antibody sequence, and demonstrated their anti-tumour activity against neuroblastoma tumour cells through a series of experiments. These PG3-GD2-CAR-T cells provide a new strategy for targeted treatment of neuroblastoma.
Footnotes
Summary Points
We successfully established a novel third-generation chimeric antigen receptor (CAR) that could specifically target human GD2 including ICOS and 4-1BB co-stimulatory domains, named PG3-GD2-CAR. PG3-GD2-CAR was constructed on normal human peripheral blood T cells to form PG3-GD2-CAR-T cells. The in vitro cytotoxicity and secretion of cytokines were detected. The results showed that PG3-GD2-CAR-T cells had better target specificity and cytotoxicity. we demonstrated that PG3-GD2-CAR-T cells significantly inhibited disease progression in mouse xenograft models of neuroblastoma cells, with minimal effects on peripheral blood cells. PG3-GD2-CAR-T cells provide a new strategy for targeted treatment of neuroblastoma.
Animal welfare
The present study followed Soochow University guidelines for humane animal treatment and complied with relevant legislation.
Author Contributions
Authors Lin Yang, Fengtao You, Cheng ji, Gangli An, Huimin Meng was responsible for study conception and design; authors Cheng ji,Tian Wang and Zhichao Han were responsible for acquisition of in vitro data; authors Cheng ji, Shuangshuang Fan,Binjie Sheng, Shufen Xiang, Yinyan Wang and Tingting Zhang were responsible for acquisition of in vivo data; authors Cheng ji, Fengtao You, Tingting Zhang were responsible for data analysis, and drafting and revision of the manuscript.
Data sharing statement
No clinical trial data were used in this study.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Ethics approval
Not applicable. This is a pre-clinical scientific research, not a clinical experimental project. It only uses human samples and does not conduct experiments on humans. At the same time, the human samples and animal experiments involved in this study were all carried out with the approval of the Ethics Committee of Suzhou University.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the National Key R&D Program of China (2016YFC1303403), the Natural Science Foundation of China (Grant No.81872431 and Grant No. 31471283), Priority Academic Program Development of Jiangsu Higher Education Institutions, the Collaborative Innovation Major Project (Grant No. XYXT- 2015304), the Six Talent Peaks Project in Jiangsu Province (No.SWYY-CXTD-010), the Natural Science Foundation of the Jiangsu Higher Education Institutions of China (General Program, Grant No. 19KJD320003).
Informed consent
Verbal informed consent was obtained from all subjects before the study. We carefully introduced the purpose of the experiment to the donors. After confirming that the donor understands the content of the experiment, we respected their choice and adopted informed Verbal consent.
Information pertaining to writing assistance
No writing assistance was utilized in the production of this manuscript.
Trial registration
Not applicable.