
Abstract
Best dystrophy (BD), also termed best vitelliform macular dystrophy (BVMD), is a juvenile-onset form of macular degeneration and can cause central visual loss. Unfortunately, there is no clear definite therapy for BD or improving the visual function on this progressive disease. The human induced pluripotent stem cell (iPSC) system has been recently applied as an effective tool for genetic consultation and chemical drug screening. In this study, we developed patient-specific induced pluripotent stem cells (BD-iPSCs) from BD patient-derived dental pulp stromal cells and then differentiated BD-iPSCs into retinal pigment epithelial cells (BD-RPEs). BD-RPEs were used as an expandable platform for in vitro candidate drug screening. Compared with unaffected sibling-derived iPSC-derived RPE cells (Ctrl-RPEs), BD-RPEs exhibited typical RPE-specific markers with a lower expression of the tight junction protein ZO-1 and Bestrophin-1 (BEST1), as well as reduced phagocytic capabilities. Notably, among all candidate drugs, curcumin was the most effective for upregulating both the BEST1 and ZO-1 genes in BD-RPEs. Using the iPSC-based drug-screening platform, we further found that curcumin can significantly improve the mRNA expression levels of Best gene in BD-iPSC-derived RPEs. Importantly, we demonstrated that curcumin-loaded PLGA nanoparticles (Cur-NPs) were efficiently internalized by BD-RPEs. The Cur-NPs-based controlled release formulation further increased the expression of ZO-1 and Bestrophin-1, and promoted the function of phagocytosis and voltage-dependent calcium channels in BD-iPSC-derived RPEs. We further demonstrated that Cur-NPs enhanced the expression of antioxidant enzymes with a decrease in intracellular ROS production and hydrogen peroxide-induced oxidative stress. Collectively, these data supported that Cur-NPs provide a potential cytoprotective effect by regulating the anti-oxidative abilities of degenerated RPEs. In addition, the application of patient-specific iPSCs provides an effective platform for drug screening and personalized medicine in incurable diseases.
Introduction
Best disease (BD), also known as best vitelliform macular dystrophy (BVMD), is characterized as a juvenile-onset form of retinal macular degeneration which can cause macular degeneration and loss of central visual capability. Clinically, BD is usually a bilateral abnormality in both adolescent and adults. It is characterized by retinal detachments filled with fluid or debris, and lipofuscin accumulation within the retinal pigment epithelium (RPE). The human bestrophin-1 gene (BEST1) encodes a protein “human bestrophin-1” which is greatly expressed in RPE. More than 120 distinct mutations in BEST1 have been found to exist, which have been identified in multiple retinal degeneration disorders, especially in BD or vitelliform macular degeneration. Functionally, human BEST1 protein has been proved to be a chloride channel which can be activated by Ca2+; most mutations causing disease in BEST1 are point mutations which lead to protein structure impairment and channel dysfunction. Therefore, decoding and understanding the structure of human BEST1 channels is worthwhile in both biological and biomedical fields.
Electrophysiological exams reveal a light peak reduction in electrooculogram (EOG) and a normal clinical electroretinogram (ERG) 1,2 . The gene responsible for BD is BEST1, which encodes the RPE-expressed protein bestrophin-1. Bestrophin-1, which consists of a 585-amino-acid transmembrane protein, is located at or close to the basolateral membrane of the RPE. This protein has been reported to act as a Ca2+-activated Cl- channel. BEST1 gene mutation may lead to the loss of anion channel function that depresses the light peak on EOG 3,4 . Bestrophin-1 also changes the activity of L type Ca2+ channels in RPE cells, suggesting that it regulates the entry of Ca2+ within RPE cells 5 . Studies showed that mutations of bestrophin-1 in the putative transmembrane domains of a murine ortholog alter its anion permeability 5,6 .
To date, in addition to supportive treatment, there is no definite cure for BD. Studies that focus on the pathophysiology of BD have been limited due to the lack of an in vitro disease model. Therefore, to develop efficient therapeutics, a detailed understanding of the pathophysiology of BD at the cellular level is essential. Human induced pluripotent stem cells (iPSCs) offer a non-invasive means to obtain specific cell types from patients with targeted diseases, and to link knowledge between clinical and bench studies 7 . Furthermore, patient-specific iPSCs from monogenic mutations have great potential for applications in modeling disease phenotypes, screening candidate drugs, and cell replacement therapies 8,9 . Together, this system provides the possibility of identifying and optimizing molecules/drugs which could lead to tight control in self-renewal, and lineage specification of iPSCs, iPSC-derived specific-lineage cells or organ tissues, as well as their functional maturation.
Recently, the development and advancement in the field of iPSCs have attracted much attention in the field of regeneration medicine and ophthalmology. The great potential and induced pluripotency have been applied in an attempt to generate multiple somatic cells, or even more complicated tissues. For example, the differentiation and generation of human photoreceptor and RPE have been reported. The capability to produce plentiful functional patient-specific retinal cells from iPSCs provides an unprecedented platform to explicate disease mechanisms and access new therapeutic candidates. Because of the unsolved puzzle of the disease-causing mechanism of BD, the search for and foundation of a disease model utilizing iPSC technology could be beneficial for clarifying the elemental questions about the biology of the disease, as well as for building up a biological toolkit to support drug discovery and toxicology verification.
Curcumin, a natural plant extract from Curcuma longa L, has been widely used in traditional Chinese medicine and in the food industry 10 . Curcumin has been reported to have various biological and pharmacological activities, including anticancer, anti-inflammatory, and antioxidant properties 11 . In addition, curcumin has been demonstrated to ameliorate the progress of macular degeneration via reducing light- and oxidant stress-induced cell death within the RPE cells, and to promote the trafficking of accumulated proteins in retinal cells 12 –14 . We utilized poly lactic-co-glycolic acid (PLGA) for the synthesis of nanoparticle-encapsulated curcumin (Cur-NPs) in order to develop a sustained-release formulation. Herein, we conducted a study with detailed genetic analysis and complete ophthalmological examinations in five members of a single family, two of whom were diagnosed with BD based upon their abnormal phenotypes and genotypes. iPSCs were generated from one of the patients with BD, and one unaffected sibling was used as a normal control. In order to gain further insight into the cellular mechanisms, a platform of personalized medicine was established via reprogramming BD patient-derived dental pulp cells into iPSCs with three reprogramming factors: Oct4, Sox2, and Klf4. Patient-specific RPE cells were differentiated from BD-iPSCs and were used as the in vitro platform for pathogenesis modeling and drug screening.
Materials and Methods
Human Dental Pulp Cells
Dental pulp cells (DPCs) from a patient with BD and an unaffected sibling were obtained from the cell bank of BIONET Corp. with the written informed consent of their parents. This study is approved by the Internal Research Board of Taipei Veterans General Hospital. Human BD DPCs and unaffected human DPCs were maintained in DMEM medium supplemented with 20% fetal bovine serum (FBS, Thermo Fisher Scientific, Waltham, MA, USA), MEM non-essential amino acids (NEAA, Thermo Fisher Scientific), L-glutamine (Thermo Fisher Scientific), and penicillin/streptomycin (Thermo Fisher Scientific) at 37°C with 5% CO2.
Human iPSC Generation and Culture
The human iPSC lines derived from one BD patient and one unaffected sibling were maintained in iPSC medium using established methods that were described in detail elsewhere 4,12,15 . Briefly, the iPSCs were reprogrammed by transduction of retroviral vectors encoding three transcription factors (Oct-4/Sox2/Klf4; OSK). At least 20 clones were established. The selected clones of iPSCs had been stably passed to >20 passages with high pluripotency. Therefore, N11 and BD01 iPSCs were selected and used in this study. Undifferentiated iPSCs were grown on mouse embryonic fibroblast (MEF) feeder layers in iPSCs medium containing DMEM/F12 (1:1) (Thermo Fisher Scientific), 20% KnockOut™ Serum Replacement (KSR) (Thermo Fisher Scientific), 1% MEM NEAA, 100 ng/ml basic fibroblast growth factor (bFGF) (R&D Systems, Minneapolis, MN, USA), 1 mM L-glutamine (Thermo Fisher Scientific) and 0.1 mM β-ME (Sigma-Aldrich, St. Louis, MO, USA). Morphologically identifiable differentiated cells were mechanically removed, and cells were passaged every 3–5 days.
Differentiation of Retinal Pigment Epithelial
For retinal differentiation, iPSC colonies were treated with 10 μM Y-27632 (STEMCELL Technologies Inc. Cambridge, MA, USA) for 1 h, and dissociated into clumps (5–10 cells per clump) with 0.25% trypsin (Thermo Fisher Scientific) and 0.1 mg/ml collagenase IV (STEMCELL Technologies Inc.) in PBS containing 1 mM CaCl2 and 20% KSR. Feeders were removed by incubation of the iPSC suspension on a gelatin-coated dish for 1 h. iPS cell clumps, at a density of 8.8 × 102 clumps/ml, were incubated in a non-adhesive MPC-treated dish (Nunc) in DMEM-F-12 supplemented with 0.1 mM 2-mercaptoethanol, 0.1 mM NEAA, 2 mM L-glutamine, and 20% KSR for 3 days, in 20% KSR-containing ES differentiation medium (GMEM, 0.1 mM NEAA, 1 mM pyruvate, and 0.1 mM 2-mercaptoethanol) for 3 days, then in 15% KSR-containing ES differentiation medium for 9 days, and finally in 10% KSR-containing ES differentiation medium for 6 days. Y-27632 (10 μM) was added for the first 15 days of suspension culture. CKI-7 (5 μM, Abcam, Cambridge, MA, USA) and SB-431542 (5 μM, Santa Cruz Biotechnology, Inc. Dallas, TX, USA) were added to the medium for 21 days during suspension culture. The medium was changed every 3 days. Formed cell aggregates were then re-plated en bloc on poly-D-lysine-laminin-fibronectin-coated eight-well culture slides (BD Biocoat, Franklin Lakes, NJ, USA) at a density of 15–20 aggregates/cm2. In the adherent cultures, cells were incubated in 10% KSR-containing ES differentiation medium. For photoreceptor differentiation, SFEB/CS-treated differentiated cells were further incubated in the photoreceptor differentiation medium [GMEM, 5% KSR, 0.1 mM NEAA, 1 mM pyruvate, 0.1 mM 2-mercaptoethanol, N2 supplement, (Thermo Fisher Scientific), 100 nM retinoic acid (Sigma-Aldrich), 100 μM taurine (Sigma-Aldrich), and 50 units/ml penicillin, 50 μg/ml streptomycin] for 50 days. The medium was changed on a daily basis.
Immunofluorescence
The living cells and spheres were fixed in 4% paraformaldehyde, permeabilized in 0.1% Triton X-100, and blocked in 5% normal goat serum–PBS. Cells were incubated with primary antibodies, including Oct-3/4 (sc-8630, 1:200), NANOG (sc-81961, 1:200), Tira-1-60 (sc-17320; 1:500) (Santa Cruz Biotechnology) and Tira-1-81 (clone 44, 1:200) (BD Biosciences, San Jose, CA, USA). After being washed three times in PBS, the cells were incubated with goat anti-mouse or secondary antibodies conjugated with FITC (green) (Abcam) or PE (red) (Abcam). DAPI was used as nuclear stain (blue) (Abcam). Images were obtained using fluorescent microscopy and a digital camera.
Quantitative PCR and RT-PCR for Marker Genes
Reverse transcription reactions were performed using SuperScript III reverse transcription (Invitrogen, St. Louis, MO, USA). cDNA was used in the following quantitative PCR (qPCR) and RT-PCR. qPCR was performed with Power SYBR Green PCR Master Mix (Applied Biosystems, St. Louis, MO, USA) according to manufacturer’s instructions. Signals were detected with 7900HT Fast Real-Time PCR system (Applied Biosystems).
In Vitro Differentiation
For in vitro differentiation, iPSCs were dispersed into small clumps using dispase (Sigma-Aldrich; 1 mg/ml for 30 min) and transferred onto ultra-low attachment plates (Corning Inc., Corning, NY, USA) for embryoid body (EB) formation. The medium was changed daily for 4 days using the same medium as for routine hESC culture. EBs were then transferred onto 0.1% gelatin-coated culture dishes with the FBS-containing medium. The medium was changed every 2 days for 20 days.
Phagocytosis Assay
Cells were incubated in medium containing Cy3-conjugated 1 μm polystyrene microspheres at a concentration of 1.0 × 108 beads/ml for 6 h at 37°C. For visualization of F-actin, the cells were stained with Alexa-Fluor-488-conjugated Phalloidin (Molecular Probes, Waltham, MA USA). The fluorescence signal was observed with a laser scanning confocal microscope.
Western Blot Assay
Western blot analysis has long been a widely used analysis technique in multiple fields of proteomics research, for example, identification and quantification of proteins, survey of protein–protein interactions, and probing post-translational proteins modifications. Western blot procedures typically contain the whole process from protein extraction, solubilization, and size exclusion/separation through SDS-PAGE, membrane transfer and subsequent antibody recognition, and radiation/fluorescence detection.
For the Western blot assay, proteins were extracted from cells and subjected to Western blot analysis. Samples were boiled at 95°C for 5 min and separated by 10% SDS-PAGE. The proteins were wet-transferred to Hybond-ECL nitrocellulose paper (Amersham, Arlington Heights, IL, USA). The following primary antibodies were used: mouse anti-iNOS (Chemicon, Temecula, CA, USA); and mouse anti-α-tubulin (Chemicon). Immunoreactive protein bands were detected by the ECL detection system (Amersham Biosciences Co., Piscataway, NJ, USA). A precision protein ladder was employed as molecular weight marker.
Preparation of Cur-NPs
Cur-NPs were prepared by the emulsion evaporation method 16 . Briefly, 50 mg of PLGA (719889, Sigma-Aldrich) and 5 mg of curcumin (Sigma-Aldrich) were dissolved in 7.5 ml acetone (9006-03, J.T. Baker, Radnor, PA, USA) as an oil phase and then added to 60 ml of an aqueous phase containing 0.1% of polyvinyl alcohol (360627, Sigma-Aldrich). The mixture was homogenized at 16,000 rpm for 15 min and then stirred at 500 rpm overnight to evaporate the organic solvent in the fume hood. The mixture was sterilized using a 0.22 µm filter and then stored at 4°C until further use. The morphology of Cur-NPs was characterized using transmission electron microscope (TEM, JEM-2000EXII, JEOL, Tokyo, Japan). The Cur-NPs solution was carefully dropped on 400 mesh carbon-coated copper TEM grid. After 15 min, the grid was tapped with filter paper to remove the excess water followed by staining with 1% phosphotungstic acid (P4006, Sigma-Aldrich) for 20 min. The samples were allowed to air dry for 24 h and then observed under TEM.
The particle size and polydispersity index (PDI) of the Cur-NPs was determined using a dynamic light scattering Particle Size Analyzer (90Plus, BIC, Holtsville, NY, USA). The zeta potential was measured using a zeta-potential analyzer (90Plus, BIC). Briefly, 10 ml of the Cur-NPs was diluted 100-fold with distilled water. The particle size, PDI, and zeta potential were analyzed by particle size and zeta-potential analyzer.
In Vitro Release from Cur-NPs
The release of curcumin from PLGA NPs was determined using a dialysis membrane method. 10 μM of Cur-NPs solution were added in a dialysis bag (1-0150-18, Orange, USA) and immersed in 10 ml of release medium containing 5 ml of both PBS and ethanol. The samples were incubated at 37°C and placed on an orbital shaker at 100 rpm. At pre-determined interval, 200 μl of sample was collected and 200 μl of fresh release medium was then replenished. The release of curcumin from NPs was determined using enzyme-linked immunosorbent assay (ELISA, Sunrise Remote, Tecan, Morrisville, NC, USA) reader at the wavelength of 420 nm. The standard curve of curcumin was measured using Tecan’s Infinite® M1000 spectrophotometer at the wavelength of 420 nm. The curcumin concentration of each sample was calculated using a linear standard curve.
MTT Assay
For evaluation of cell survival, cells were seeded on 24-well plates at a density of 5×104 cells/well, followed by the addition of methyl thiazol tetrazolium (MTT; Sigma-Aldrich) at the end of cell culture. The amount of MTT formazan product was determined using a microplate reader at an absorbance of 560 nm (SpectraMax 250, Molecular Devices, Sunnyvale, CA, USA).
Determination of Intracellular Reactive Oxygen Species (ROS) Production
The measurement of intracellular reactive oxygen species (ROS) production was done by using a peroxide/ hydroperoxide probe 20,70-dichlorofluorescein diacetate (DCFH-DA; Molecular Probes, Eugene, OR, USA). In brief, cells were incubated with 5 mmol/L DCFH-DA in culture medium for 30 min at 37°C, followed by washing with PBS and detected by flow cytometry analysis.
Statistical Analysis
The results are expressed as mean ± SD. Statistical analyses were performed using the t-test for comparing two groups, and one-way or two-way ANOVA, followed by Bonferroni’s test, was used to detect differences among three or more groups. Results were considered statistically significant at p<0.05.
Results
Generation of iPSCs from BD Patient
Dental pulp-derived stromal cells (DP-SCs) were isolated and cultured from dental pulp tissue following our previous protocol 17 . To further generate iPSCs, DP-SCs obtained from the BD patient and an unaffected sibling were transfected with retroviral vectors encoding Oct4, Sox2, and Klf4. During the reprogramming process, these cells progressively formed colonies with increasing size. These colonies were cultivated on MEF feeder cells in KSR-based media. They were positive for alkaline phosphate staining and morphologically indistinguishable from human embryonic stem cells (ESCs) (Fig. 1A). To improve clinical utility and prevent MEF contamination, these reprogrammed iPSCs were transferred to feeder-free culture in CSTI-8 medium without KSR supplementation as previously described 17 . In this serum- and feeder-free system, both unaffected sibling-derived iPSCs (Ctrl-iPSC) and BD-derived iPSCs (BD-iPSC) could be stably passaged for at least 20 passages. In the reprogramming process, we selected the clumps that encompassed iPSCs-like cells for continuous subculture and named them as different clones. As shown by RT-PCR, clones of Ctrl-iPSC and BD-iPSC expressed various genes which were identical to the H9 human ESC lines such as: Oct4, Sox2, klf4, Nanog, REX, DPPA2, and GDF3 (Fig. 1B). Immunofluorescence confirmed the strong expression of stemness-associated factors, including Oct4, Nanog, Tra1-60, and Tra1-81 in iPSCs at 20th passage (Fig. 1C). There was no significant difference in the expression of pluripotent genes between BD-iPSCs and iPSCs from the unaffected sibling and BD patient. This result suggested that bestrophin-1 deficiency did not hinder the efficiency of reprogramming and maintaining pluripotency. Thus, bestrophin-1 did not play a role that affects the generation of iPSCs.

Generation and characterization of iPSCs derived from a patient with Best disease and an unaffected sibling. (A) Phase-contrast photomicrograph and alkaline phosphatase activity of undifferentiated control iPSCs (ctrl-iPSCs) and Best disease iPSCs (BD-iPSCs). Scale bar = 200 μm. (B) The RT-PCR results indicated an embryonic stem cell (ESC)-like gene expression pattern in representative colonies of iPSCs. Dental pulp-derived stromal cells (DR-SC) were used as a negative control, and human ES-H9 cells as a positive control. (C) Immunofluorescence staining demonstrated the expression of pluripotency markers (OCT4, TRA-1-61, Nanog, and TRA-1-81) in undifferentiated iPSCs from patient and unaffected sibling. Nuclei were counterstained with DAPI (blue). Scale bar = 100 μm. (D) Immunofluorescence staining demonstrated markers of all three germ layers that were expressed in spontaneously differentiated iPSCs. Nuclei were counterstained with DAPI (blue). Scale bar = 20 μm. (E) Sequence analysis revealed the existence of specific Best mutations in the BD-iPSC lines.
Using differentiation protocols for tridermal lineages, both 20th-passage Ctrl-iPSC-derived EBs and BD-iPSC-derived EBs (data not shown) could be induced to differentiate into neuron-like cells (ectoderm lineage), smooth muscle cells (mesoderm lineage), and hepatocyte-like cells (endoderm lineage). Each lineage was exhibited by specific canonical markers, including neuronal nuclear, glycoprotein of fetal liver, nestin, smooth muscle specific cytoskeleton (SMA), and alpha-fetoprotein (AFP) (Fig. 1D). In the process of differentiation, we demonstrated that both Ctrl- and BD-iPSCs were able to differentiate into three germ layers.
Lastly, DNA sequencing analysis demonstrated the mutation of the BEST1 gene in BD-iPSCs but not in Ctrl-iPSCs (Fig. 1E), confirming that BD-iPSCs retained the BEST1 gene mutation. In summary, these results revealed that both BD-iPSCs and Ctrl-iPSCs exhibited pluripotent properties and were capable of multi-lineage differentiation.
Comparison of BD-RPEs and Ctrl-RPEs
Because RPE has been closely linked to the pathogenesis of BD, we examined whether RPEs derived from BD-iPSCs could exhibit the typical pathophysiological features of BD. Elegant work done by Osakada et al. has demonstrated that human ESCs and iPSCs could be used as alternative sources for differentiated retinal progenitors, RPE cells, and photoreceptors 18 . We employed the culture protocol described by Osakada et al. with brief modifications to differentiate BD-iPSCs into RPEs (Fig. 2A). Under progressive stimulations, tight hexoagonal pigmented patches appeared and increased over time. After 30–40 days of differentiation, these patches were collected for culture. As shown in Fig. 2B, both Ctrl-iPSCs and BD-iPSCs underwent RPE-specific morphological changes with pigmentation indicating that they have differentiated into RPE-like cells.
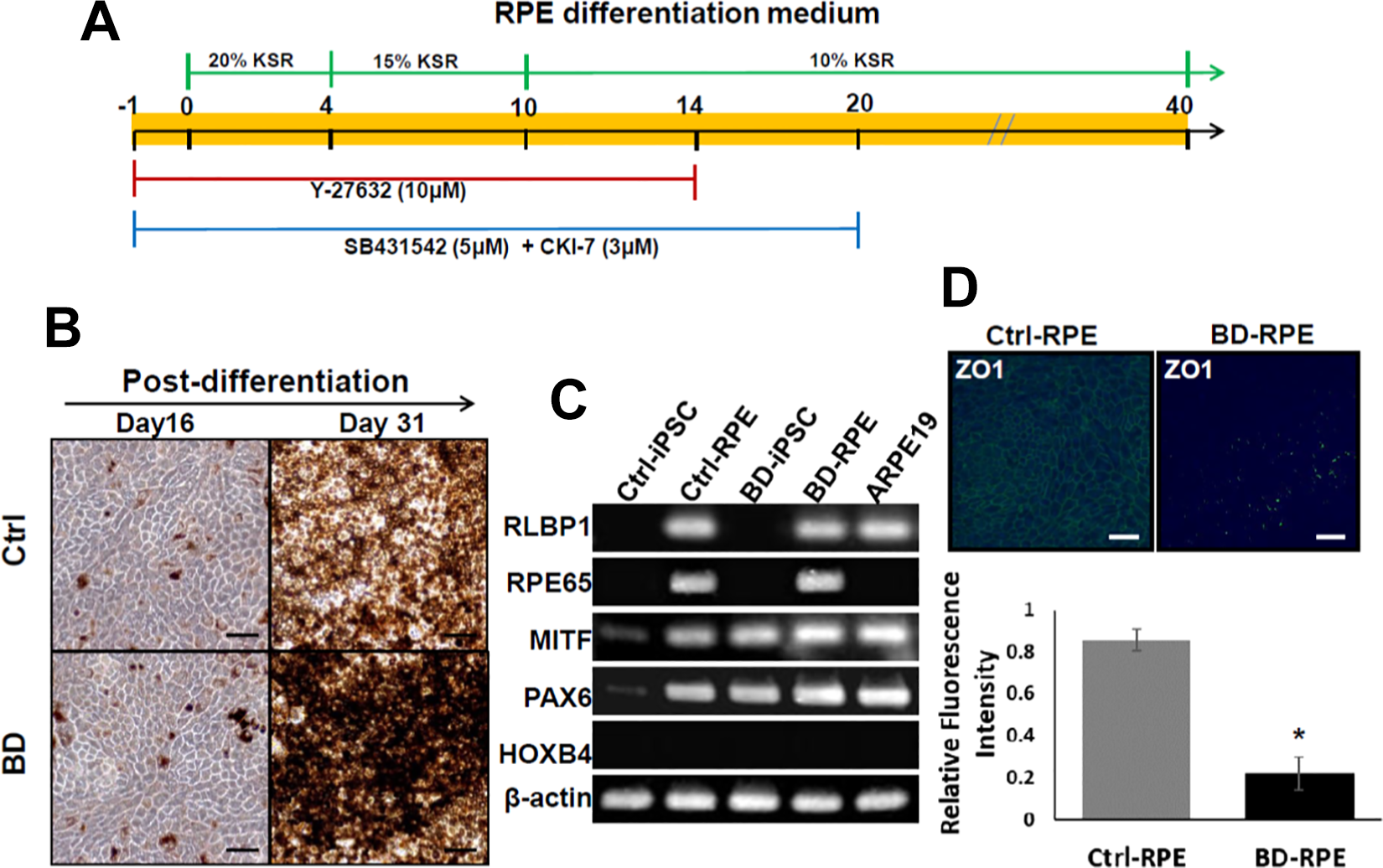
(A) Schematic diagram of the culture protocol for differentiation of BD-iPSCs into RPEs. (B) Ctrl-iPSCs and BD-iPSCs both underwent RPE-specific morphological changes with pigmentation at day 16 and 31. (C) RT-PCR showed that Ctrl-iPSCs and BD-iPSCs were competent to be differentiated into pigmented cells with typical RPE characteristics. ARPE19 cells did not express RPE65. BD-RPEs had weaker RPE65 expression compared with Ctrl-RPEs. (D) Immunofluorescence assays showed that BD-RPEs had significant lower ZO-1 expression than Ctrl-RPEs.
RT-PCR confirmed the expression of several RPE-specific markers, including RLBP1, RPE65, MITF, and PAX6 in both Ctrl-RPEs and BD-RPEs (Fig. 2C). The results showed that both Ctrl-iPSCs and BD-iPSCs could be differentiated into pigmented cells with typical RPE characteristics. ARPE19, an immortalized RPE cell line derived from the normal eyes of a 19-year-old male, currently used as an in vitro model for human RPE cells 19 and serving as a control cell line in this study, also expressed RPE markers except RPE65. In comparison to Ctrl-RPE, BD-RPE expressed weaker RPE65, which indicates the possibility of a diminishing effect of aberrant BEST1 on visual cycle-associated proteins.
The expression levels of tight junction protein ZO-1 were compared between the Ctrl-RPEs and BD-RPEs. Immunofluorescence assays showed that BD-RPEs had significant lower ZO-1 expression than Ctrl-RPEs (Fig. 2D).
BD-RPEs as a Drug-Screening Platform
A platform for screening candidate drugs in vitro was established using patient-specific BD-RPEs to model the pathophysiological features of BD (Fig. 3A). In comparison with normal RPE cells, BD-RPEs showed lower expression of ZO-1 (Fig. 3B, upper) and CACNA1C (Fig. 3B, lower). Several dietary retinal protection supplements and natural antioxidant compounds, including β-carotene (Carotent, 10 μM), lutein (5 μM), retinoic acid (RA, 10 μM), forskolin (5 μM), isoproterenol (Isuprel, 200 μM), resveratrols (10 μM), vitamin C (Vit C, 10 μM), curcumin (10 μM), and Q10 (20 μM) were selected (Fig. 3C and 3D). Among all candidate drugs, only curcumin largely restored the mRNA expression of BEST1, ZO1, and CACNA1C.
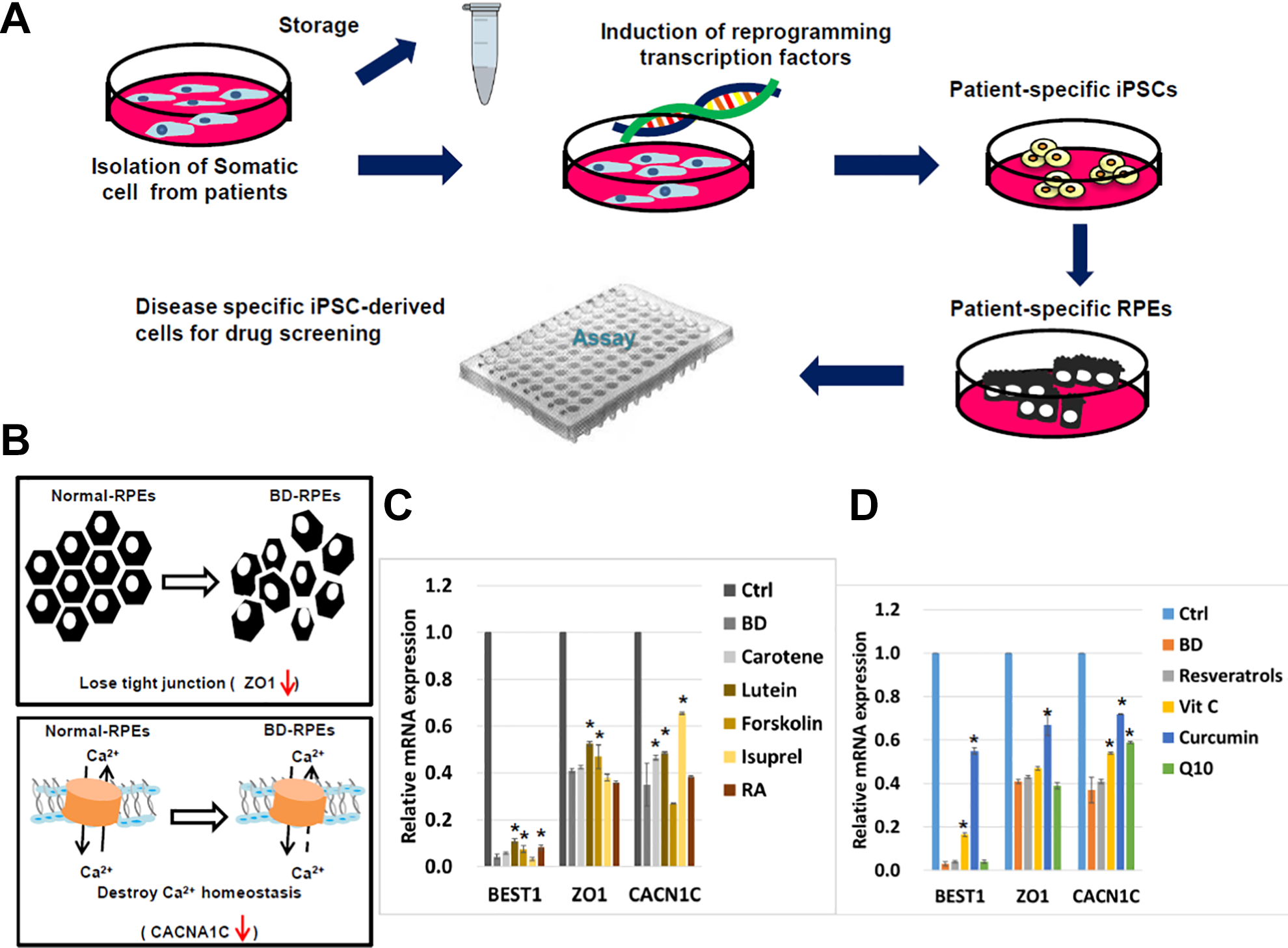
(A) Patient-specific iPSCs are a promising disease model for drug screening. (B) In comparison with normal RPE cells, less expression of ZO1 and CACNA1C expression in BD-RPEs indicating the loss function of tight junction and Ca2+ homeostasis. (C&D) mRNA expression level of BEST1, ZO1, and CACNA1C in normal RPE cells and BD-RPEs after treated with β-carotene (Carotent, 10 μM), lutein (5 μM), retinoic acid (RA, 10 μM), forskolin (5 μM), isoproterenol (Isuprel, 200 μM), resveratrols (10 μM), vitamin C (Vit C, 10 μM), curcumin (10 μM), and Q10 (20 μM).
Curcumin-encapsulation in PLGA Nanoparticles
Nanoparticle (NP) technology could be applied in targeted drug delivery based upon its advantage of small volume (generally <100 nm), bioavailability, and its ability to contain biodegradable materials to improve uptake. The application of curcumin has been limited due to its hydrophobic nature and non-solubility in water. Therefore, an alternative method to overcome this limitation is to prepare curcumin NPs with water solubility. PLGA, a material with excellent biocompatibility and biodegradability, was used for the synthesis of curcumin NPs (Cur-NPs). Curcumin encapsulated in NPs completely dissolved in aqueous solution without aggregation (Fig. 4A). The surface morphology of the nanosphere-encapsulated curcumin under TEM scanning demonstrated a spherical and smooth particle (Fig. 4B). The size, PDI, zeta potential, encapsulation efficiency, and concentration of Cur-NPs were 145.0 ± 6.0 nm, 0.040 ± 0.022, –23.8 ± 2.6 mV, 46.2 ± 1.7%, and 312.27 ± 11.80 μM, respectively. The results showed that the size of Cur-NPs is consistent with low PDI. Encapsulation of curcumin in PLGA NPs may increase the size distribution. Curcumin is an uncharged electrically neutral compound which may increase the zeta potential of PLGA NPs. The final concentration of Cur-NPs was 312.27 ± 11.80 μM.
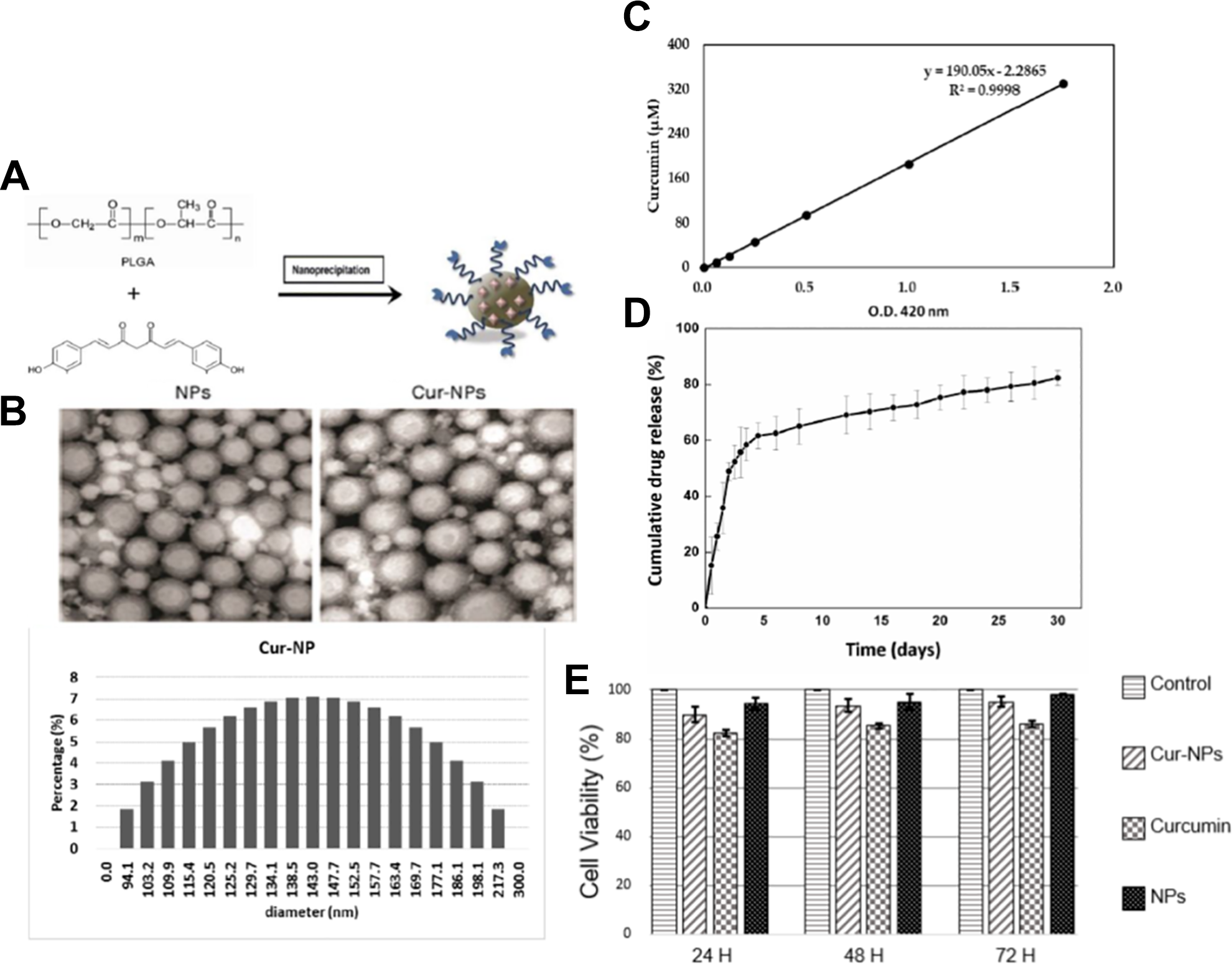
Development of PLGA-based sustained releasing therapeutic vehicles. (A) Character of curcumin encapsulated PLGA vehicle (Cur-NPs). (B) Transmission electron microscopy (TEM) scanning of Cur-NPs. (C) In vitro release kinetic profile and cumulative percentage of curcumin released from of Cur-NPs. (D) Colorimetric WST-1 assay showed that RPE proliferation is not hindered after treating with Cur-NPs and regrowth in regular medium for 72 h.
The in vitro release kinetic profile and cumulative percentage of curcumin released from Cur-NPs are shown in Fig. 4C. On the first day, 40% of the curcumin was released from Cur-NPs, which then increased to 80% on day 3 and 90% on day 5. Afterwards, curcumin released from Cur-NPs was sustained at approximately 80% until day 10. Drug release from Cur-NPs displays multiple release phases, including initial burst release, lag phase, and zero-order release 20 . The initial burst release is regulated by diffusion of the surface- and pore-associated drug; the lag phase and zero-order release are controlled by polymer erosion combined with diffusion. Furthermore, previous research demonstrated that the low molecular weight PLGA NPs exhibited diffusion-controlled release 21 . Accordingly, the sustained release of curcumin from NPs prepared with low molecular weight PLGA is controlled by diffusion. Sustained release of the drug in a delivery system is an important property closely related to therapeutic pharmacokinetics and efficacy.
Cell viability and proliferation are essential regarding cytotoxicity in the biotechnological criteria. To evaluate whether the Cur-NPs possess potential cytotoxicity against RPE, a colorimetric WST-1 assay was used to assess the cell viability and proliferation. Colorimetric WST-1 assay showed that RPE proliferation is not hindered after treatment with Cur-NPs and regrowth in regular medium for 72 h (Fig. 4D). Regarding biosafety, Cur-NPs showed nominal cytotoxicity.
Restoration of Anti-oxidative and Retino-physiological Functions in BD-RPE by Cur-NPs
The cytoprotective effects of Cur-NPs on restoration of functions of BD-RPEs were investigated. The expression of the BEST1 gene was significantly lower in BD patient-derived iPSCs and RPEs compared with Ctrl-iPSCs and Ctrl-RPEs (Fig. 5A). After treatment with 10 μM Cur-NPs for 72 h, the results of Western blotting revealed that Cur-NPs substantially increased the protein expression level of BEST1 in BD-RPEs (Fig. 5B).

Identification of the therapeutic effect of Cur-NPs on BD-iPSCs. (A) qRT-PCR of BEST1 was quantified relative to the expression levels in the indicated groups. (*p<0.05, compared with the unaffected sibling). The data illustrate the mean ± SD of three independent experiments. (B) qRT-PCR and western blotting with the representative level revealed that Cur-NPs substantially increased BEST1 levels in BD-RPEs. (C&D) Immunofluorescence images of quantification of BEST1 and ZO1 expression in BD-RPEs after Cur-NPs treatment. (E) Laser scanning confocal microscopy and quantitative fluorescence imaging of phagocytosis by Ctrl-RPEs and BD-RPEs (lower). Nuclei were counterstained with DAPI (blue). Scale bar = 50 μm for ZO1; 10 μm for phagocytosis. The data shown here are the mean ± SD of three independent experiments. *p<0.05, compared with nontreated NPs.
The immunofluorescence assays confirmed that treatment with Cur-NPs largely restored the expression of BEST1 and ZO-1 (Fig. 5C and 5D). The results of labeled latex bead phagocytosis assays revealed an improvement in the phagocytic ability of BD-RPEs after treatment with Cur-NPs (Fig. 5E).
The antioxidant activity of Cur-NPs was measured by qRT-PCR. The expression of genes involved in the regulation of cellular stress and inflammatory processes, such as HO-1, VEGF, SOD2, GPX1, and TRFR, was significantly decreased in BD-RPEs compared with Ctrl-RPEs. After supplementation with Cur-NPs, the basal expression mRNA levels of these genes were significantly upregulated (Fig. 6A).

The therapeutic effect of Cur-NPs on oxidative stress in BD-RPE. (A) qRT-PCR was used to quantify the relative amounts of antioxidant genes in the indicated groups. *p<0.05 compared with Ctrl-RPE; #p<0.05 compared with BD-RPE. The data shown here are the mean ± SD of three independent experiments. (B) Quantification of ROS levels in BD-RPEs after Cur-NPs treatment. (C&D) The survival rate (% of control) of BD-RPEs after Cur-NPs treatment was determined. *p<0.05; #p<0.05. The data shown here are the mean ± SD of three independent experiments.
The results detected by flow cytometry showed that H2O2-induced ROS generation was higher in BD-RPEs than in Ctrl-RPEs (data not shown). Both 100 μM and 200 μM sizes of H2O2 were selected to mimic the in vivo reality and to determine the clearance effect of Cur-NPs. After administration of Cur-NPs, a decrease in H2O2-induced ROS and maintained low-level intracellular ROS in BD-RPEs were noted (Fig. 6B).
Protective effects of Cur-NPs against acute or chronic oxidative stress were tested by evaluating the survival rate (% of control) of BD-RPEs after Cur-NPs treatment. In the acute oxidative stress group, which was treated with H2O2 for 30 min, the results showed upregulation of stress-correlated proteins. In contrast, the chronic oxidative stress group, treated by H2O2 for 6 h, showed numerous inflammatory factors and severely induced cell death (Fig. 6C). In addition, administration of Cur-NPs demonstrated the rescue effect of BD-RPEs from H2O2-induced cell death (Fig. 6C and 6D).
Discussion
Human iPSCs have the ability to propagate infinitely and to differentiate into various cell types of the human body 22 . In addition, the advantages of these cells, such as high consistency, purity, and expandability, could be applied in drug screening, toxicity testing, and the development of personalized medicine 23,24 . Patient-specific iPSCs and their differentiated progenies have provided new opportunities for recapitulating particular individualized disease phenotypes, in which in vitro studies could be conducted to understand the mechanisms and pathogenesis of degenerative diseases 25,26 . Furthermore, personalized medicine in the prevention, diagnosis, and treatment of specific disease could be customized for each patient based on their own genetic profile 27 . In this study, we demonstrated that reprogrammed human dental pulp-derived stromal cells from a BD patient could be developed into BD-iPSCs and be stably passaged for at least 50 passages with good pluripotency and the ability for tridermal differentiation. Dental pulp has been reported as a source of multipotent progenitors and pluripotent stem cells 15,28 . It could be isolated from human exfoliated deciduous and permanent teeth 29 . Due to its differentiation potential and easy accessibility, the number of studies focusing on dental pulp in regenerative medicine has gradually increased 30,31 . The gene responsible for BD is BEST1, which encodes the RPE-specific protein bestrophin-1. Since human BEST1 is a regulator of calcium, its deficiency has been shown to elicit a cascade series that impacts either the morphology or function of RPE cells. Calcium is a major cofactor for most adhesion proteins, particularly in RPE cells that rely on the assistance of tight junctions 32,33 . Therefore, we generated BD-RPEs and Ctrl-RPEs by inducing the differentiation of their corresponding iPSCs. At day 16 and 31 post-differentiation, Ctrl-iPSCs and BD-iPSCs both underwent RPE-specific morphological changes with pigmentation. RT-PCR confirmed the typical expression of RPE-specific genes, including PAX6, MITF, RLBP1, and RPE65 in both Ctrl-RPEs and BD-RPEs. PAX6 is the marker of retinal progenitor 34 , and MITF is an important indicator of pre-RPE 35 , and RLBP1 36 and RPE65 37 are both cellular RPE markers. The BD-RPEs were able to replicate multiple pathophysiological features of BD, including bestrophin-1 deficiency, downregulation of CACNA1C expression that disrupted intracellular calcium homeostasis, loss of the tight junction protein ZO-1, and reduction in the RPE’s phagocytic ability.
Pomares et al. reported that mRNA expression level of BEST1 gene not only reflects disease progression but also disease severity 38 . Accordingly, we evaluated the mRNA expression levels in BD-RPEs after treatment with various candidate drugs. In addition to the BEST1 gene, the mRNA expression level of tight junction (ZO-1) and alpha-1 subunit of a voltage-dependent calcium channel (CACNA1C) were also investigated to reflect the abnormal gene expression in BD. Taken together, our data suggested that BD-RPEs provide a powerful tool for modeling BD phenotypes and could be used as a high-throughput and expandable platform for in vitro drug screening.
Curcumin has been used as a popular traditional Chinese medicine because of its diverse and broad biological activities, including antioxidant, anticancer, and neuro-protective effects 39,40 . In addition, curcumin has also been reported to have remarkable efficacy in rescuing neurodegenerative disorders 39,41 and RPE apoptosis from light or oxidative stress 12,13 . These treatment effects of curcumin were attributed to the upregulation of oxidative stress defense enzymes 13 and the activation of several cellular regulatory proteins that inhibit cellular inflammatory responses 12,13,42 . Curcumin has also been reported as an epigenetic mediator. It was found to regulate histone deacetylase, histone acetyltransferase, DNA methyltransferase I, and microRNA 43,44 . Through epigenetic modulation, curcumin is capable of mediating gene expression 45 . In the present study, administration of curcumin significantly restored the mRNA expression and protein amount of the BEST1 gene in BD-RPEs, suggesting that curcumin may serve as an epigenetic effector to upregulate the expression of BEST1 in BD-RPEs.
Oxidative stress-induced damage is a major factor in the progression of retinal degeneration, including age-related macular degeneration and other hereditary retinal diseases 12,13 . Notably, the RPE comprises part of the blood–retina barrier, which is highly susceptible to ROS 13,46 . We found that incubation of BD-RPEs with curcumin suppressed H2O2-induced ROS production and ameliorated rates of cell death. Although the detailed mechanisms of the curcumin effect that rescues RPE functions in BD-RPEs are not fully understood, our data indicated that a remarkable ROS scavenging effect was involved in this curcumin-mediated cytoprotection in BD-RPEs.
Nevertheless, several disadvantages, including low water solubility, poor absorption, rapid metabolism, and fast systemic elimination, have limited the bioavailability of curcumin 40 . In recent years, several novel drug delivery systems have been widely discussed to improve the bioavailability of hydrophobic active pharmaceutical ingredients. These delivery systems include NPs, lipid-based vesicles, and micelles 47 –50 . Therefore, we developed a Cur-NPs-based controlled release formulation and demonstrated its effect on increased expression of ZO-1 and Bestrophin-1, which promoted the function of phagocytosis and voltage-dependent calcium channels in BD-RPEs. Our data also showed that Cur-NPs could reduce intracellular ROS production and decrease oxidative stress induced by hydrogen peroxide. Sustained release of a high level of curcumin in the culture BD-RPEs could be detected for up to 10 days. To the best of our knowledge, this is the first report of the delivery of curcumin as NPs in patient-derived RPE cells from patients with BVMD. This intervention study demonstrates a novel application of iPSC-based personalized medicine.
In conclusion, our results showed that patient-specific iPSCs, such as BD-derived iPSCs, can act as an efficient drug-screening tool, leading to further personalized medicine. The cytoprotective effect of Cur-NPs provides therapeutic potential in regulating the anti-oxidative abilities in degenerative retinal diseases.
Footnotes
Acknowledgments
We deeply thank Dr. Chien, Ke-Hung, Dr. Chuang, Jen-Hua, and Dr. Wang, Mong-Lien for supporting this study and providing valuable comments.
Ethical Approval
All of the procedures and protocols for this study were approved by the Institutional Review Board of Taipei Veterans General Hospital.
Statement of Human and Animal Rights
All of the procedures involving human subjects were conducted in compliance with the Declaration of Helsinki and ARVO statement. Laboratory animals are not applicable for this article.
Statement of Informed Consent
Informed consent was obtained from the human subjects used in this article.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research and/or authorship of this article: This study was funded by VGH-NTUH Joint Research Program (VN106-02, VN107-16, VN108 (Dr. Yang, Chang-Hao & Dr. Chiou, Shih-Hwa), the “Center for Intelligent Drug Systems and Smart Bio-devices (IDS2B)” from The Featured Areas Research Center Program within the framework of the Higher Education Sprout Project-NCTU by the Ministry of Education (MOE) in Taiwan. Ministry of Science and Technology (MOST), Academia Sinica and MOST (MOST 104-0210-01-09-02, 105-0210-01-13-01, 106-0210-01-15-02, and 107-0210-01-19-01), Department of Health and Welfare of Taiwan (Health Cancer Center Research of Excellence & Clinical Trial Center of Excellence MOHW105-TDUB-211-134003, MOHW105-TDU-B-211-133017, MOHW106-TDU-B-211-113001, and MOHW107-TDU-B-211-123001), NRPB Human iPSC Alliance-Core Service (MOST 105-2325-B-010-005), VGH, TSGH, NDMC, AS Joint Research Program (VTA105-V1-5-1, VTA107-V1-5-1, VTA108), VGH, NTUH Joint Research Program (VN106-02, VN107-16, VN108), and National Health Research Institutes (NHRIEX106-10621BI, and NHRI-EX107-10621BI), Taiwan. This work was also financially supported by the “Cancer Progression Research Center, National Yang-Ming University.”