
Abstract
Neuronal apoptosis is regarded as one of the most important pathophysiological changes of intracerebral hemorrhagic (ICH) stroke—a major public health problem that leads to high mortality rates and functional dependency. Mitogen-and stress-activated kinase (MSK) 1 is implicated in various biological functions in different cell types, including proliferation, tumorigenesis and responses to stress. Our previous study showed that MSK1 phosphorylation (p-MSK1) is related to the regulation of LPS-induced astrocytic inflammation, and possibly acts as a negative regulator of inflammation. In this study, we identified a specific interaction between MSK1 and MRKβ (MLK-related kinase)—a member of the MAPK pathway—during neuronal apoptosis. In an ICH rat model, western blotting and immunohistochemical analysis revealed that both MRKβ and phosphorylation of MSK1 (p-MSK1 Ser376) were significantly upregulated in cells surrounding the hematoma. Triple-immunofluorescent labeling demonstrated the co-localization of MRKβ and p-MSK1 in neurons, but not astrocytes. Furthermore, MRKβ was partially transported into the nucleus, and interacted with p-MSK1 in hemin-treated neurons. Immunoprecipitation showed that MRKβ and p-MSK1 exhibited an enhanced interaction during the pathophysiology process. Utilizing small interfering RNAs to knockdown MRKβ or MSK1, we verified that MSK1 Ser376 is a phosphorylation site targeted by MRKβ. We also observed that the phosphorylation of NF-κB p65 at Ser276 was mediated by the MRKβ-p-MSK1 complex. Furthermore, it was found that the neuronal apoptosis marker, active caspase-3, was co-localized with MRKβ and p-MSK1. In addition, flow cytometry analysis revealed that knockdown of MRKβ or MSK1 specifically resulted in increased neuronal apoptosis, which suggested that the MRKβ-p-MSK1 complex might exert a neuroprotective function against ICH-induced neuronal apoptosis. Taken together, our data suggest that MRKβ translocated into the nucleus and phosphorylated MSK1 to protect neurons via phosphorylation of p65—a subunit of nuclear factor κB.
Introduction
Intracerebral hemorrhagic (ICH) stroke, a devastating disease caused mainly by hypertension and arteriosclerosis, affects approximately 2 million people worldwide each year 1 . Due to medical advances, the mortality rate associated with ICH has declined; however, the neurological impairments of survivors will either persist or develop later in life 2 . Accumulating evidence has showed that ICH can lead to neurological impairment due to immediate central nervous system (CNS) tissue disruption owing to dynamic hematoma expansion. Besides, ICH can also result in secondary injury by complex mechanisms, such as inflammation, ischemia, brain edema, and oxidative stress triggered by detrimental cascades, leading to further lesion and disability 3,4 . After ICH, neurons undergo wholesale cellular pathophysiologic processes involving the release of inflammatory cytokines, apoptosis, necrotic cell death, and autophagy 4,5 . Among these, neuronal apoptosis is considered one of the most crucial devastating events in the long-term pathological consequences of ICH, resulting in various neurological deficits 5 . Although there is no debate about the existence of neuronal apoptosis in ICH stroke, the regulatory mechanisms of apoptosis after ICH stroke remain controversial. To this end, we utilized the rat intracerebral hemorrhage and cellular apoptosis models to explore the possible signaling pathways involved in neuronal apoptosis.
Mitogen-and stress-activated kinase (MSK) 1 is a nuclear-localized protein activated in cells downstream of the extracellular signal-regulated kinase (ERK) or p38 MAPK pathways 6,7 . As a key regulator of transcription, activated MSK1 regulates diverse cellular responses, including the inflammatory response, cell proliferation, cell differentiation, and apoptosis, by subsequently phosphorylating their respective nuclear substrates 7 –9 . Several studies have identified that MSK1 can phosphorylate subunits of NF-kB and CREB, both of which are key transcription factors targeting genes involved in different neuronal processes 10 –12 . We recently reported that MSK1 phosphorylation (p-MSK1) is associated with the regulation of LPS-induced astrocytic inflammation, and possibly acts as a negative regulator of inflammation 13 . Furthermore, we also found that p-MSK1 was expressed abundantly in neurons 13 . However, the biological significance and the potential mechanisms of MSK1 in the neuronal pathophysiological process remain unclear.
MSK1 is activated via a complex series of phosphorylation and auto-phosphorylation reactions, depending on the stimulus and cell types used 6 . It was thought that Ser-360 and Thr-581 phosphorylation by ERK or p38 were essential for the catalytic activity of MSK1, and, thus, for the phosphorylation of MSK1 substrates 14 . However, studies showed that MSK1 is not a direct phosphorylated substrate of ERK and p38 MAPK in adult rat cardiac myocytes 15 . Thus, we hypothesize that MSK1 is activated on a certain site via an as yet unidentified activator.
It was reported that, aside from p38 and ERK, MRKβ can activate MSK1 through direct phosphorylation of several important sites 16 . MRKβ, also called ZAK, is an upstream kinase of the MAPK cascade and can activate the JNK and NF-kB pathways 17 . A previous study showed that the expression of MRKβ was associated with the apoptosis of tumor cells, including cutaneous squamous cell carcinoma and hepatoma-cells 18,19 . Moreover, in human 293 T cells and HeLa cells, MRKβ potentially interacts with, and phosphorylates, MSK1 at multiple sites 16 . Indeed, the biological significance and the underlying mechanisms of MRKβ and MSK1 in neuronal apoptosis after ICH stroke remain unclear.
Nuclear factor κB (NF-κB) is a key regulator of transcription of a variety of genes expressed in response to injury or inflammatory stimuli. When NF-κB is released from its inhibitor IκB, it translocates to the nucleus to activate expression of various target genes 20,21 . A growing body of recent data indicates that ERKs and p38 are implicated in NF-κB transactivation, and that this action is probably mediated by MSK1 12,22 . In addition, some researchers observed that members of the caspase family and the Bcl-2 family, which are strongly associated with cellular apoptosis, were mediated by NF-κB in some cancer cells 23 . However, whether the activation of NF-κB was mediated by the MRKβ-MSK1 complex in neurons after stroke has not been explored.
The purpose of the current study was to identify whether MRKβ activates MSK1 through phosphorylation during the process of neuronal apoptosis. Our data suggest that MRKβ nuclear transportation and the subsequent activation of the MRKβ-p-MSK1-NF-κB axis may magnify upstream signals, eventually acting as a novel neuronal protective mechanism against intracranial hemorrhagic injury.
Materials and Methods
Intracerebral Hemorrhagic Stroke Model in Rats
All surgical experiments and postoperative animal care were carried out in accordance with the guidelines prescribed by the Chinese National Experimental Animals for Medical Purposes Committee, Jiangsu Branch. Adult male Sprague-Dawley rats (weighing 220–250 g) were obtained from Nantong University Laboratory Animal Center and housed in a temperature-controlled (21°C) and 60–80% humidity environment. Surgeries were performed in mice according to a previous published paper with minor modifications 24 . The rats were anesthetized intraperitoneally with chloral hydrate (10% solution) and positioned in a stereotactic frame. Then, autologous freely dripping blood (100 μL) was collected by cutting the tail tip. Autologous whole blood was injected stereotaxically into the right basal ganglia (coordinates 0.2 mm anterior, 5.5 mm ventral, 3.5 mm lateral to the bregma) at a rate of 10 μL/min by a microinfusion pump. The sham-operated group had only a needle insertion. The sterile syringe was left in place for over 10 min, removed slowly, and the scalp was closed with sutures afterward. Surgical animals (n = 4 per time point) were sacrificed to extract the protein for western blot analysis at 3, 6, and 12 h at 1, 3, 5, and 7 days following ICH. The sham-operated rats (n = 3) were sacrificed on the second day. Additional experimental animals (n = 6 per time point) for sections were sacrificed at each time point for histopathological analysis.
Behavioral Testing Procedures
Forelimb placing test
The rats were held by the torso, which allowed the forelimb to hang free. Intact rats would quickly put the forelimb ipsilateral onto the countertop. Placing of the forelimb contralateral to the injury may be impaired based on the extent of injury. During the tests, each rat (six rats for per time point) was measured 10 times in each forelimb. The percentage of trials in which the rat placed the left forelimb in response to the vibrissae stimulation was calculated.
Corner turn test
The rats were allowed to proceed into a corner, the angle of which was 30°C. To exit the corner, the rats could turn either to the left or the right randomly, and this was recorded. This was repeated 10–15 times, with at least 30 s between trials, and the percentage of right turns was calculated as the corner turn score. Only turns involving full rearing along either wall were included. Ventral tucks or horizontal turns were excluded.
Cell Cultures and Treatment
Primary cortical neurons were obtained from dissociated cortex of embryonic day-18 (E18) rats. In brief, the cerebral cortices were mechanically dissected and digested with trypsin. After digestion, the precipitate was re-suspended, and the isolated cells were seeded in the culture medium, supplemented with neurobasal medium containing 5% fetal bovine serum, 5% horse serum. Cells were maintained at 37°C in a humidified 5% CO2 incubator. Half of the culture medium was changed every 2–3 days. Culture neuronal cells were used at day 8. To mimic hemorrhagic conditions in vitro, cells were exposed to hemin (Sigma) in 50 μmol/L for the indicated times.
Western Blotting Analysis
The protein was separated by SDS-PAGE, transferred onto PVDF membranes (Bio-Rad, Hercules, CA, USA), and immunoblotted with antibodies. The primary antibodies used were as follows: rabbit anti-MRKβ (1:1000; Abcam, Cambridge, UK), mouse anti-MSK1 (1:1000; Abcam), mouse anti-p-MSK1 (1:500; Abcam), rabbit anti-Bax (1:1000; Abcam), and mouse anti-caspase-3 (1:500; Abcam). Internal controls included mouse anti- GAPDH (1:1000; Santa Cruz Biotechnology, Santa Cruz, CA, USA) or mouse anti-β-actin (1:1000; Sigma-Aldrich, St. Louis, MO, USA). After incubation with HRP-conjugated secondary antibodies (anti-rabbit or anti-mouse; 1:2,000; Promega, Madison, WI, USA) for 2 h at 37°C, proteins were visualized using an ECL detection system (ECL, Pierce Company, Waltham, MA, USA). The optical density of the band was assessed using Image J.
Sections and Immunohistochemistry
At the defined survival times, rats were anesthetized and perfused with 0.9% saline and 4% paraformaldehyde separately through the ascending aorta. The brains were removed and fixed in the same fixative overnight, and then the fixative was replaced with 20% sucrose for 2–3 days, and then 30% sucrose for 3–4 days. The tissues were embedded in OCT compound. Then, 5-μm coronary frozen sections were prepared using a freezing microtome. All sections were stored at –20°C until use. For immunohistochemistry (IHC), sections were first blocked with confining liquid consisting of 10% donkey serum, 0.3% Triton X-100, and 1% bovine serum albumin (BSA) for 2 h at RT. Then, the sections were incubated overnight at 4°C with anti-MRKβ antibody (rabbit, 1:100; Abcam) or anti-p-MSK1 antibody (1:100; Abcam) followed by incubation with a biotinylated secondary antibody. Staining was visualized using DAB chromogen. The stained sections were mounted with a Leica light microscope (Leica, Wetzlar, Germany), coupled to a Leica DFC300FX microscope camera.
Immunofluorescent Staining
Rat brain sections were prepared as described above. Cultured neurons were fixed with 4% paraformaldehyde and permeabilized for 3 min with 1% Triton X-100 at room temperature (RT). Brain sections or neuronal cultures were first incubated in blocking solution containing 3% BSA and 0.1% TritonX-100 for 2 h at RT. Then, the sections were labeled with the primary antibodies against the protein of interest overnight at 4°C, and lastly incubated with the specific FITC- and TRITC-conjugated secondary antibodies (1:200; Abcam) for 2 h at RT. Nuclei were stained with DAPI (1:2000, Sigma). The immunoreactive complexes were visualized and analyzed with a Leica TCSSP8 confocal system. Quantitative analysis of the extent of colocalization was performed with ImageJ software.
Co-immunoprecipitation
Tissues surrounding hematomas (2 mm) from the ICH or neurons treated with hemin were lysed in 500 μl RIPA buffer (50 mM Tris-HCl, 1% NP-40, 1% PMSF, 1 mM EDTA-Na, 0.02% sodium azide, 0.1% SDS, 0.5% sodium deoxycholate, 150 mM NaCl). The lysates were centrifuged at 13,000 rpm for 20 min at 4°C to collect the supernatants. The lysate supernatant was incubated with protein G (Santa Cruz Biotechnology, Santa Cruz, CA, USA) at 4°C for 2 h followed by incubated with either anti-MRKβ or anti-p-MSK1 antibodies at 4°C overnight. The precipitates were isolated by centrifugation, washed three times with lysis buffer, and immune complexes were eluted by heating at 100°C in loading buffer and used for immunoblotting analysis.
Nuclear and Plasma Separation
The nucleus and plasma separation of neurons was performed according to the nucleus and plasma separation kit (Sangon Biotech Cooperation, Shanghai, China) instructions. After separation, the samples were kept at –80°C until analysis.
Small Interfering RNA Transfection
Three small interfering RNAs (siRNAs) targeting the MRKβ and MSK1 genes were designed and synthesized, and the most effective siRNA (as identified by Western blotting) was applied for further experiments. The sequences of three siMRKβ were made to target the sequence: 5’-GATCCGCCAGTGGTTAGATACTCTG-3’, 5’-TTGAGCTCATGTCGTCTCTCGGTGC-3’, and 5’-AGCTTAAGCCTCTCTTCCATAACCA-3’. The sequences of three siMSK1 were made to target the sequence: 5’-AATCGCATAGCGTATGCCGTT-3’, 5’-AGCCACATGCACGATGTAGGA-3’, and 5’-CATCAGCCACGAGACATCAAC-3’. Scrambled RNA oligonucleotides were used as controls. All plasmids were verified by DNA sequencing analyses. Cells were transfected using Lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA), as suggested by the manufacturer. The transfection efficiency was measured by Western blotting. The concentration of siRNA used was 20 μM.
Analysis of Apoptosis by Flow Cytometry
The apoptosis analysis was conducted following the protocol of Annexin V/PI apoptosis detection kit (Calibur, BD Biosciences, Franklin Lakes, NJ, USA). The neurons were treated with hemin for 12 h before they were analyzed by FACS. After treatment, up to 2–9 × 105 cells for each sample were analyzed using CellQuest software.
Statistical Analysis
Stata 7.0 software and GraphPad Prism were used for statistical analysis and data plotting. Quantitative values were presented as means ± SD. Differences of the mean value between groups were calculated using the student’s t test. One-way ANOVA followed by Tukey’s post hoc multiple-comparison tests were used. The experiments were repeated at least three times. P < 0.05 was considered statistically significant.
Results
Spatiotemporal Changes of MRKβ and p-MSK1 Expression after ICH
To assess acute and chronic changes in sensorimotor function and plasticity in the ICH model, two behavioral tests were used: a forelimb placing test and a corner turn test 25 . Rats that received an intracerebral infusion of autologous whole blood had significant forelimb placing deficits and significant increases in the percentage of right (ipsiversive) turns compared with sham controls from days 1 through 14 (Fig. 1A, B). This result showed that the ICH group exhibited apparent neurological deficits in the two tests over the 1st day (*P < 0.05). Western blotting was performed to investigate the temporal levels of MRKβ and p-MSK1 in brain tissues around the hematoma at various time points after ICH. Compared with the sham group, MRKβ protein level increased gradually at 3 h following the ICH operation, and reached maximal levels at day 3. In addition, p-MSK1 (S376) expression was also upregulated in a time-dependent manner. However, total MSK1 expression did not change among the different time groups (Fig. 1C, D). IHC staining with anti-MRKβ and anti-p-MSK1 antibodies on transverse cryosections of brain tissues was performed to examine brain histological changes 3 days after ICH. As shown in Fig. 2, the sham-operated group showed relatively low MRKβ-positive and p-MSK1-positive staining, similar to the level of the contralateral group. On the contrary, the number of MRKβ- and p-MSK1-positive cells in the ipsilateral group was significantly upregulated close to the hematoma (Fig. 2E, F, M, N), which was consistent with the Western blotting data.
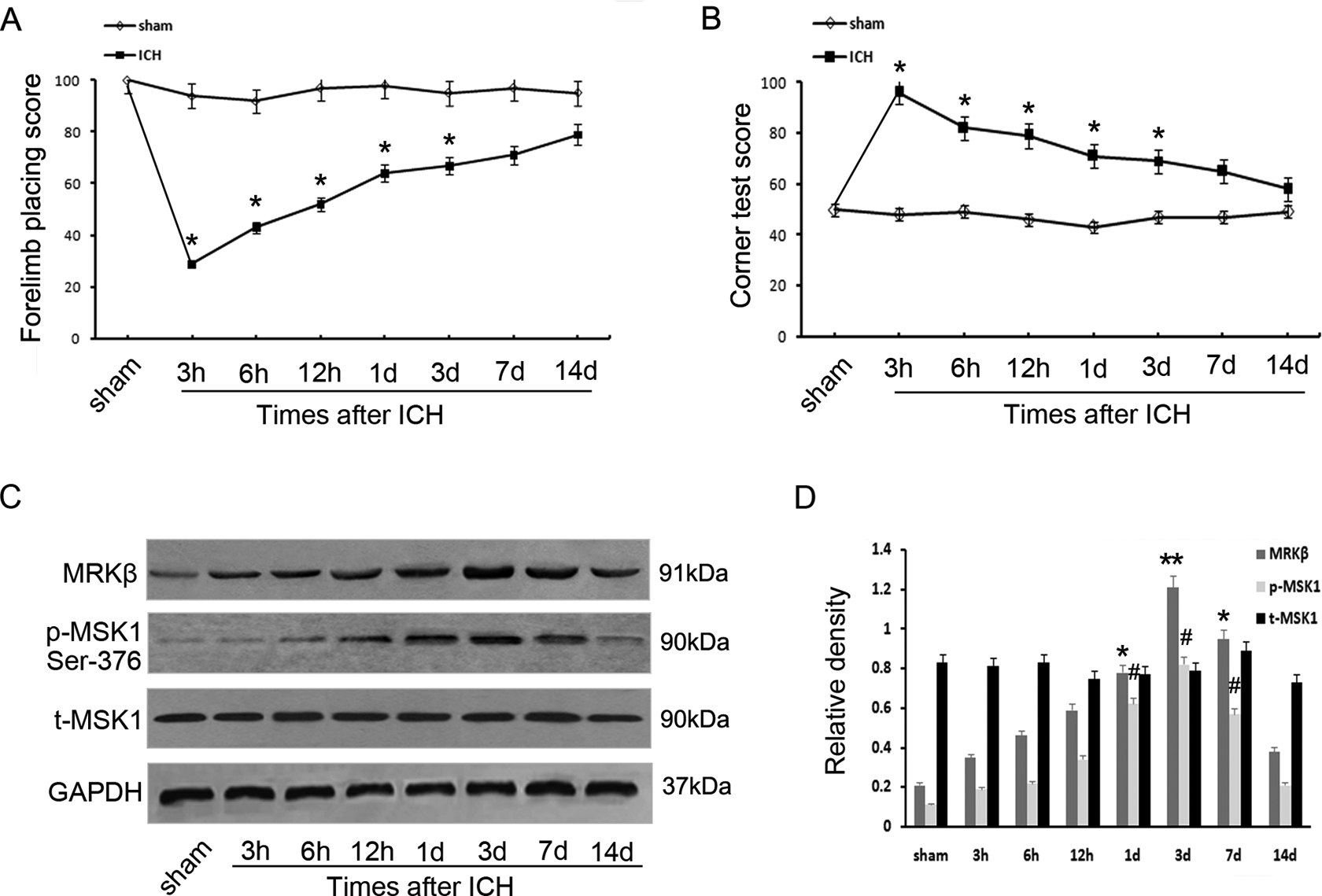
Time-dependent variation in MRKβ/MSK1/p-MSK1 protein expression following ICH in rats. Forelimb placing scores (A) and Corner turn testing (B) were displayed at various time points following ICH. (C) Western blot was employed to detect the protein levels of MRKβ, MSK1, and p-MSK1 adjacent to the hematoma at different survival times. (D) The relative protein contents of MRKβ, MSK1, and p-MSK1 were calculated by densitometry analysis, and the data were normalized to corresponding GAPDH levels. The behavioral testing experiments were repeated at least three times.

Expression and distribution of MRKβ and p-MSK1 immunohistochemistry surrounding the hematoma. (A, B) MRKβ was relatively low in the sham group. At day 3 after ICH, the number and intensity of MRKβ positive cells had significantly increased in the ipsilateral group (E, F) compared with the sham control group (A, B) or the contralateral group (C, D). (G) Quantitative analysis of MRKβ-positive cells per square millimeter. (H, I) p-MSK1 staining was detected in sham group. (M, N) Significant increase of p-MSK1 staining was detected 3 days post-ICH in the ipsilateral group (M, N) compared with the sham control group (H, I) or the contralateral group (J, K). (O) Quantitative analysis of p-MSK1-positive cells per square millimeter. *Significant difference at P < 0.05 compared with sham group. Error bars represent SEM. Scale bars 50 μm (A, C, E, H, J, M), 20 μm (B, D, F, I, K, N). The experiments were repeated at least three times.
Subcellular Localization of MRKβ and p-MSK1
To further confirm the subcellular localization of MRKβ and p-MSK1 after ICH, immunofluorescence staining was performed with cell-specific markers. As shown in Fig. 3, double-labeling immunofluorescence staining showed there was no co-localization of MRKβ and GFAP in the brain tissues 3 days after ICH (Fig. 3A–C). In contrast, obvious MRKβ immunoreactivity was observed in NeuN+ cells with neuronal morphology (Fig. 3 G, H). Meanwhile, it was found that p-MSK1 was expressed widely in neurons, with a relatively low level of expression observed in astrocytes in the ICH groups (Fig. 3 D–J). Furthermore, triple-immunofluorescent labeling showed MRKβ+/p-MSK1+ staining was expressed in neurons, which indicated that MRKβ and MSK1 might interact and participate in neuronal apoptosis after ICH.
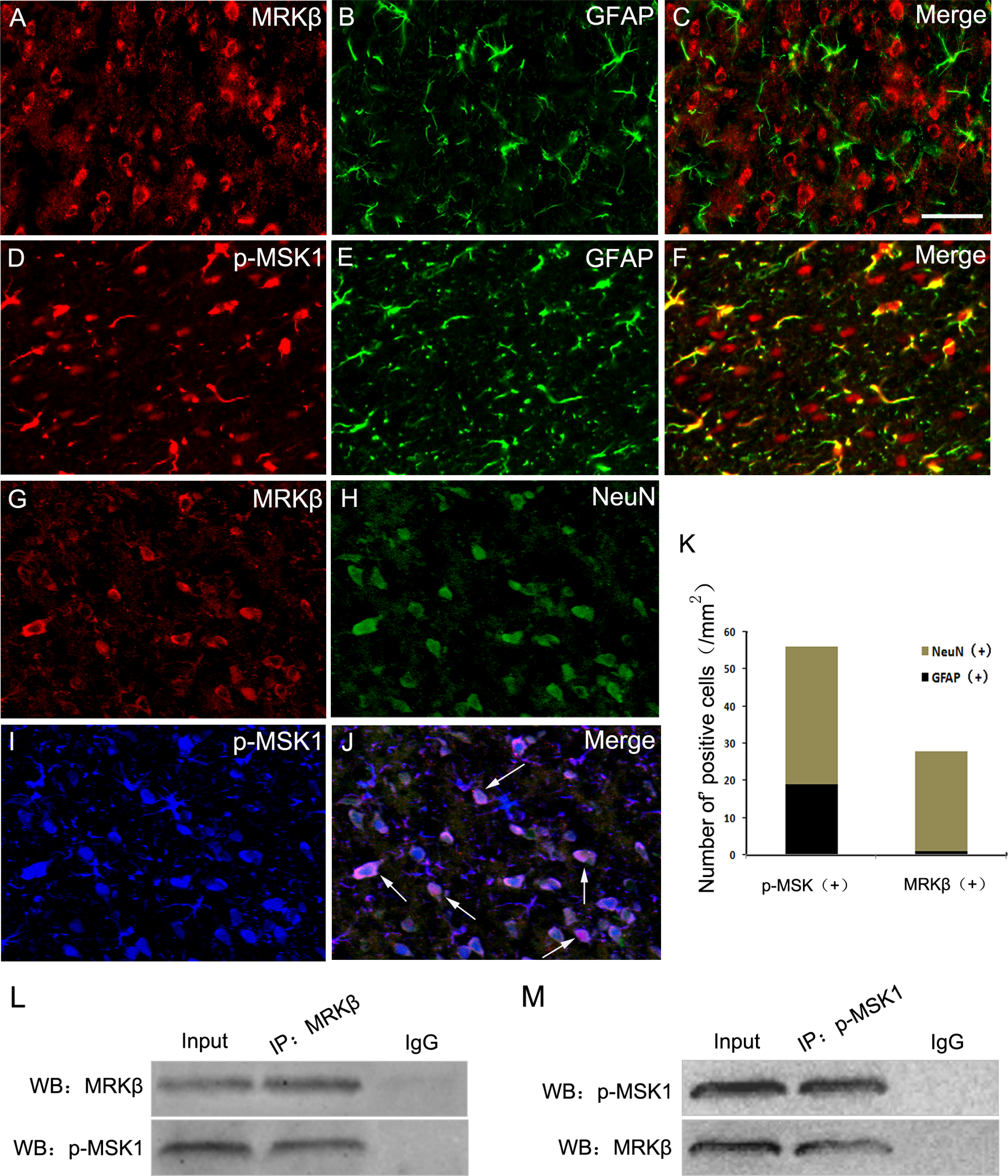
Immunofluorescence staining for MRKβ and p-MSK1 in brain. (A–C) In the rat after ICH, horizontal sections labeled with MRKβ and astrocyte marker (GFAP). (D–F) Double immunofluorescence staining for p-MSK1 and GFAP. (G–J) Triple immunofluorescence staining for MRKβ, p-MSK1 and neuronal marker (NeuN).The yellow color visualized in the merged images represents the colocalizations of p-MSK1 with GFAP (F). The purple visualized in the merged images represents the colocalizations of p-MSK1 with MRKβ and NeuN (J). Total p-MSK1+ and MRKβ+ cells were counted in the same area after ICH. Scale bars 20 μm (A–J). (L, M) Immunoprecipitation showed that the interaction of MRKβ and p-MSK1 in the experimental group 3 days after ICH.
Interaction of MRKβ and p-MSK1 in Vivo and in Vitro
To verify this possible interaction between MRKβ and p-MSK1 following ICH, an immunoprecipitation assay was performed in the tissues surrounding the hematoma at day 3 following ICH (Fig. 3 L, M). Given that MRKβ and p-MSK1 are closely involved in neuronal apoptosis, we set out to determine the possible functions of the MRKβ-p-MSK1 complex during this process. Rat primary neurons were stimulated with hemin (100 μmol/L) to establish the apoptosis model. Then, we detected the protein levels of MRKβ, p-MSK1, and t-MSK1. The levels of MRKβ and p-MSK1 increased significantly after treatment, with both peaking at 12 h. However, total MSK1 did not change (Fig. 4A, B). To further evaluate the intracellular interaction between MRKβ and p-MSK1, we exposed the neurons to hemin to mimic ICH in vitro for further investigation. Immunoprecipitation assays demonstrated that hemin enhanced the interaction of MRKβ and p-MSK1 in neurons (Fig. 4C, D). In addition, immunofluorescence staining showed the co-localization of MRKβ and p-MSK1 in neurons (Fig. 4E). Interestingly, an obvious alteration in MRKβ subcellular localization was detected by immunofluorescent staining. As shown in Fig. 4E, MRKβ expression was mainly in the cytoplasm in normal neurons, but accumulated in the nucleus 6 h after hemin treatment. Then, we further confirmed the nuclear transportation of MRKβ by Western blotting. Consistent with the immunofluorescence results, the expression of MRKβ in the nucleus increased to a maximum at 6 h post hemin treatment, and decreased gradually thereafter (Fig. 4F). Based on this result, we hypothesized that MRKβ could transfer to the nucleus, where it phosphorylates p-MSK1 and participates in neuronal apoptosis.

MRKβ nuclear transportation and interaction with p-MSK1 in hemin-treated neurons. (A, B) Western blot showing expression patterns of MRKβ, p-MSK1 and MSK1 in neurons after hemin treatment at different times. (C, D) Immunoprecipitation showing that the interaction of MRKβ and p-MSK1 improved significantly in neurons suffered from hemin compared with the sham group. (E) Immunofluorescence assay examining the subcellular distribution and colocalization of MRKβ (red) and p-MSK1 (green) in rat primary neurons following hemin stimulation. Scale bar, 10 μm. (F) Nuclear and cytoplasmic extracts of neurons were prepared as described in ‘Materials and Methods’ and examined by Western blot to determine the nuclear translocation of MRKβ stimulated by hemin. Each experiment consisted of at least three replicates per condition.
MRKβ-mediated MSK1 Phosphorylation Induced Activation of the NF-κB Pathway
Uninjured neurons were collected and the knockdown efficiency of MRKβ siRNA and MSK1 siRNA was examined by Western blotting (Fig. 5A, B). MRKβ protein expression was markedly reduced in MRKβ siRNA-transfected neurons treated with 100 μmol/L hemin for 6 h, compared with the control group. MRKβ downregulation with siRNAs also decreased the protein level of p-MSK1(Ser376) induced by hemin. However, there was no obvious decrease in total MSK1 expression among the various group (Fig. 5C), suggesting that MSK1 may possibly play biological roles after being phosphorylated by the new activator, MRKβ. However, following MSK1 knockdown, there was no significant change in MRKβ expression (Fig. 5D). These results indicate that MRKβ may regulate MSK1 expression in rat primary neurons stimulated with hemin via an upstream phosphorylation cascade. It was reported that phosphorylation of the NF-κB p65 subunit at Ser276 induced by oxidative stress is likely mediated by MSK1. Thus, to further investigate the activation of NF-κB and possible interactions with the MRKβ-p-MSK1 complex in the process of neuronal apoptosis, the main phosphorylation site of p65 (Ser276) was examined. Western blotting confirmed that hemin induced significant phosphorylation of p65 at the second amino acid residue (Ser276). Furthermore, Ser276 phosphorylation induced by hemin was significantly downregulated in neurons transfected with siRNA targeting MRKβ or MSK1 (Fig. 5C, D). Relative protein expression levels were measured and compared with β-actin to create the quantification graphs (Fig. 5E–J).

MRKβ-mediated MSK1 phosphorylation induced the activation of NF-κB pathway. (A) Western blot showed the effects of siRNAs for MRKβ on expression of MRKβ in neurons. (B) Western blot showed the effects of siRNAs for MSK1 on expression of MSK1 in neurons. (C) Effects of siRNA for MRKβ on hemin-induced expression of MRKβ, p-MSK1 (Ser376), MSK1, and p-p65 (Ser276) were detected by Western blotting. (D) Effects of siRNA for MSK1 on hemin-induced expression of MRKβ, p-MSK1 (Ser376), MSK1, and p-p65 (Ser276) were detected by Western blotting. The bar chart indicated the density of MRKβ/ MSK1/p-MSK1/p-p65 versus β-actin (E–J). The data are mean ± SEM. (n = 3, # means P < 0.05).
Regulation of MRKβ-p-MSK1 on Cell Apoptosis in Vitro
To further examine the precise role of MRKβ-p-MSK1 in the process of neuronal apoptosis following ICH, a series of experiments were performed. Western blotting analysis reflected a time-dependent increased synthesis of the activated forms of caspase-3 and Bax, both in brain tissues surrounding the hematoma and in neurons, after stimulation with hemin (Fig. 6A–D). Immunofluorescent staining was also employed to examine the distribution and co-localization of MRKβ, p-MSK1, and activated caspase-3 in the ipsilateral brain cortex. Double immunofluorescent staining showed co-localization of activated caspase-3 and NeuN in the brain cortex 1 day after traumatic brain injury, confirming neuronal apoptosis following ICH (Fig. 6K–M). Additionally, caspase-3 fluorescence appeared in many MRKβ- or p-MSK1-expressing cells at day 1 after ICH (Fig. 6E–J). Flow cytometry using an Annexin V/PI apoptosis detection kit was performed to determine the presence of apoptotic cells. Hemin stimulation caused a notable increase in neuronal apoptosis, whereas both MRKβ-siRNA and p-MSK1-siRNA remarkably induced a higher level of apoptotic cells compared with the control group (Fig. 6 N, O). These results indicated that activated MRKβ-p-MSK1 inhibited neuronal cell death and induced the development of a neuronal protective mechanism against ICH-induced apoptosis.
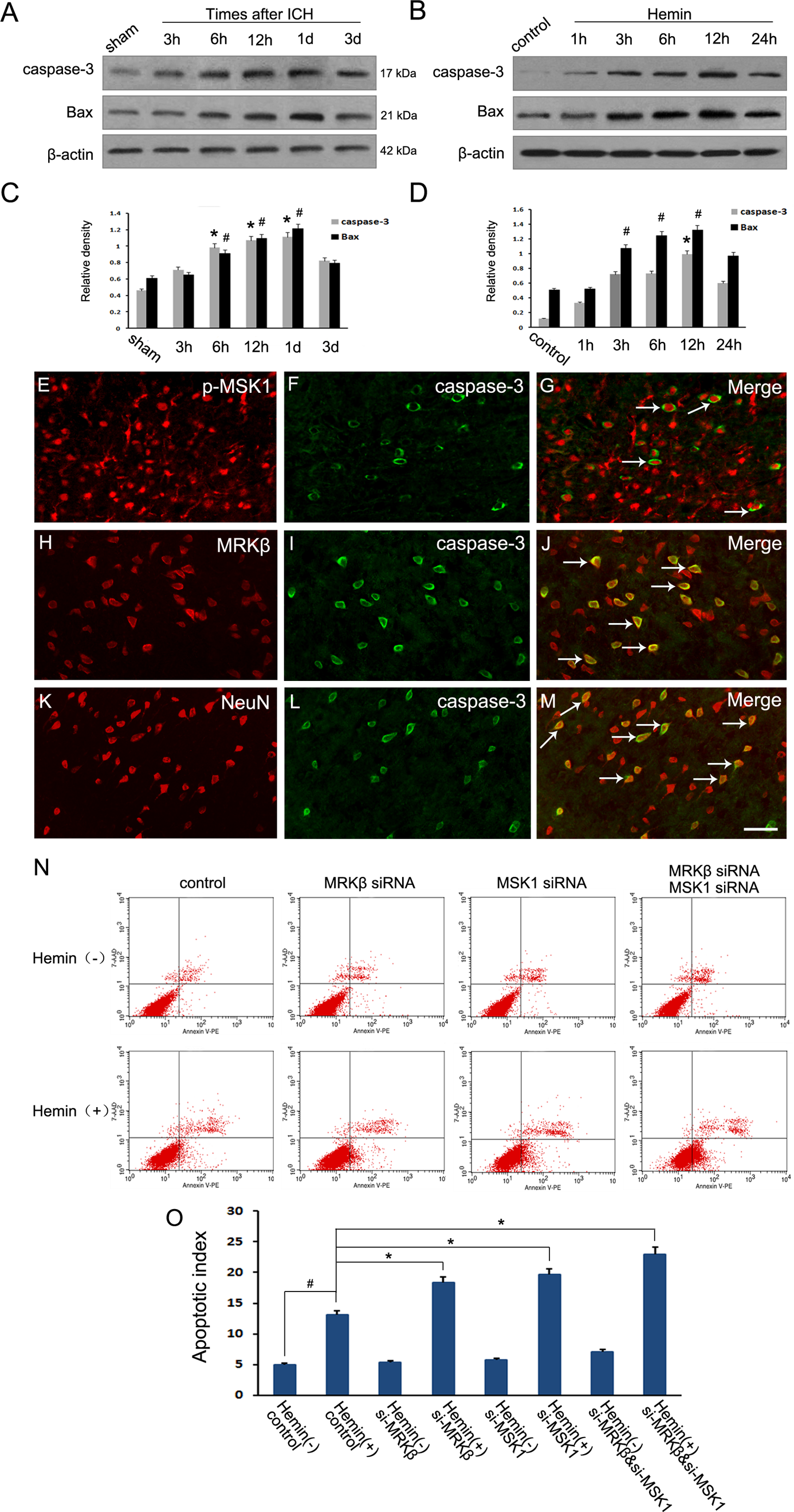
Modulations of MRKβ-p-MSK1 on neuronal apoptosis after ICH. (A, C) Western blot analysis showed the protein level of active caspase-3 and Bax increased adjacent to the hematoma after ICH. (B, D) Western blot showed active caspase-3 and Bax increased in neurons after hemin exposure. (E–M) Horizontal sections of brain labeled with active caspase-3 (Green F, I, L), NeuN (Red K), MRKβ (Red H) and p-MSK1 (Red E). The yellow color visualized in the merged images represented colocalization of active caspase-3 with NeuN, MRKβ and p-MSK1. (as the white arrows show in G, J, M). Scale bars 20 μm. (N) The apoptotic cells were determined using an Annexin V/PI apoptosis detection Kit (Sigma, St. Louis, MO, USA) for flow cytometry (Calibur, BD Biosciences, Franklin Lakes, NJ, USA), according to the manufacturer’s instructions. (O) The apoptotic index of different groups according to annexin V/PI staining. * P < 0.05, n = 3.
Discussion
Intracerebral hemorrhage is one of the most detrimental CNS diseases; ICH is associated with high morbidity and mortality and lacks effective treatment. The primary injury caused by ICH is associated with the dynamics of hematoma expansion and subsequent parallel pathological pathways such as brain edema, an intracranial inflammatory microenvironment, oxidative reactions, regional ischemia, and hypoxia, which contribute to secondary injuries 3,4 . Among these pathological changes, neuronal apoptosis exerts a vital influence on neurological impairments after ICH, involving complex proapoptotic and anti-apoptotic signals 3,26 . Therefore, gaining a better understanding of the underlying molecular and cellular mechanisms of neuronal apoptosis following ICH may ultimately improve the outcomes of cerebral-hemorrhagic stroke.
In our previous studies, we found that LPS induced the phosphorylation of MSK1 at Thr-581in cultured astrocytes. Besides, the sequential activation of the MAPK and p-MSK1 (Thr-581) pathways may possibly attenuated the production of inflammatory cytokines in LPS-treated primary astrocytes 13 . In addition to regulating astrocytic inflammatory reactions, the fluorescence results also showed that activated MSK1 was largely localized in neurons, but the underlying molecular mechanisms and biological significance remains unclear. Therefore, in this study, we further evaluated if MSK1is activated by a new upstream activator and if phosphorylation of MSK1 could activate a downstream substrate, such as NFκB, further contributing to neuronal apoptosis.
On the basis of the amount of intracranial hematoma and the predilection site in clinical scenarios, we utilized a controlled autologous blood intracerebral injection ICH model in adult rats to verify the involvement of the MRKβ-p-MSK1-p65 axis in neuronal apoptosis and ICH progression. In our study, rats suffering from ICH showed serious functional deficits, as assessed by behavioral testing. Western blot and immunohistochemical analyses indicated that both MRKβ and p-MSK1 were significantly increased in regions surrounding the hematoma after ICH. Since these two kinases were co-localized mainly in neurons, not in astrocytes, we wondered whether they were involved in neuronal apoptosis after ICH. As expected, we found that the expression patterns of caspase-3 and Bax were parallel to those of MRKβ and p-MSK1 in vivo and in vitro. Simultaneously, depletion of MRKβ or p-MSK1 by siRNA in neurons can increase hemin induced cellular apoptosis. All of the above data suggested that the MRKβ-p-MSK1 pathway can exert biological function(s) in neuronal death via the extrinsic apoptotic pathway.
Although the roles of MSK1 phosphorylation in inflammation and immunity have been studied extensively, the subsequent downstream events that result in neuronal apoptosis remain less clear. Indeed, some paradoxical conclusions about MSK1 in apoptosis have been reported recently. Some data indicated that phosphorylation of the MSK1-CREB cascade is an essential component of activity-dependent hippocampal neuronal cell death 27 . Some results identified MSK1 as a novel and direct signaling mediator of Bad phosphorylation, thereby further preventing widespread cell death in epidermal cells treated by UVB radiation 28 . Some researchers have found that heavy metal (Mn2+)-induced apoptosis of human lymphoma B cells is dependent on sequential p38-MAPK and MSK1 activation 29 . It was also reported that ischemic-preconditioning-mediated activation of the downstream target MSK-1, which, in turn, phosphorylates CREB and transmits survival signals in cardiomyocytes 30 . Because the phosphorylation of different sites in MSK1 can affect its activity, it seems necessary to evaluate the functions of the individual phosphorylation sites in the process of neuronal apoptosis.
Previous evidence has shown that the phosphorylation of MSK1 does not require additional signaling input, aside from extracellular signal-regulated kinases (ERKs) and p38 MAPK 7,29,31 . Our results suggest that, in addition to ERK1/2 and p38, MRKβ may be a novel kinase that can phosphorylate MSK1 at multiple serine sites in response to certain external stressors and stimuli. MRKβ is a member of the MAPKK kinase (MAPKKK) family of signal transduction molecules 17 . Although it was reported that knockdown of MRKβ suppressed phospho-JNK activation and apoptosis in cancer cells, not many studies have focused on the biological significance of MRKβ in the CNS, especially in neuronal apoptosis. Here, we report for the first time that MRKβ can be transported into the nucleus and directly phosphorylate MSK1 at Ser376 during the process of neuronal damage. However, other important phosphorylated sites of MSK1 (such as Ser360 and Thr581), which were reported to be phosphorylated by ERK1/2 and p38, were not identified in our repeated assays. Although p-MSK1 (Thr-581) was reported as a pro-survival kinase downstream of the p38 pathway, there is no evidence that Thr-581 is related to neuronal apoptosis 27 . Further studies are also needed to determine the activation mechanisms and biological functions of these phosphorylated sites. Considering the sequential activation of MRKβ and MSK1 induced by hemin in neurons, it is reasonable to speculate that this may further lead to the transcriptional activity of nuclear substrates that protect neurons against hemin. In our results, we found that si-MRKβ or si-MSK1 distinctly suppressed the phosphorylation level of NF-κB p65. This finding is consistent with the report that MSK1 mediates NF-κB dependent transcription through phosphorylation of the NF-κB isoform (p65) on Ser276 12,20,21 . Further, MRKβ belongs to the family of serine/threonine kinases and can activate the NF-κB pathway 17 . Activation of the NF-κB pathway may lead to enhancement of neuronal cell survival during oxidative stress, suggesting that the anti-apoptotic effects of lipoic acid in desflurane-treated neurons was mediated by NF-κB signaling 32 . The efficacy of some anticancer drugs is also due to decreased NF-κB protein expression 33 . This is because NF-κB-dependent transcription produces proteins that protect cells from apoptosis. However, further studies are necessary to explore the potential target genes of the MRKβ-p-MSK1 complex in neuronal apoptosis.
In conclusion, our results provide a novel and logical explanation for the molecular mechanism involved in neuronal apoptosis after ICH. Mechanistically, after the transcription of MRKβ is activated, MSK1 can be phosphorylated and play a neuroprotective role against hemin-induced neuronal apoptosis. We also found that this protective process against hemin in neurons is correlated with the activation NF-κB signaling. Further experiments will be necessary to identify NF-κB as the downstream gene target of the MRKβ-MSK1 phosphorylation cascade in neurons following intracerebral hemorrhage. Therefore, understanding the molecular signals of MRKβ-MSK1-NF-κB in the ICH-induced neuronal survival pathway may be helpful in designing therapeutic targets for the clinical treatment of ICH.
Footnotes
Acknowledgments
Ethical Approval
This study was approved by the Chinese National Experimental Animals for Medical Purposes Committee, Jiangsu Branch.
Statement of Human and Animal Rights
All of the experimental procedures involving animals were conducted in accordance with the Institutional Animal Guidelines of Nantong University, China and approved by the Chinese National Experimental Animals for Medical Purposes Committee, Jiangsu Branch.
Statement of Informed Consent
There are no human subjects in this article and informed consent is not applicable.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the Youth Program of National Natural Science Foundation of China (81401013, 81700696), the Project of Youth Medical Talents in Jiangsu Province (QNRC2016692), the scientific research program of Jiangsu Health Commission (H2017053) and the new clinical diagnosis and treatment technology research project (MS22016047).