
Abstract
Cerebral aneurysm growth is characterized by vessel wall frailness, although the underlying cellular mechanisms are unclear. Here, we examined the relationship between inflammatory smooth muscle cells (SMCs) and endothelial cells (ECs) in cerebral aneurysms, including the mechanisms underlying inflammatory SMC-induced changes in ECs. Five saccular cerebral aneurysms were collected and five temporal artery samples were used as controls. Cells and cytokines were detected by immunohistochemistry and TUNEL (transferase dUTP nick end labeling) assays performed to evaluate apoptosis. Human umbilical vein endothelial cells (HUVECs) were seeded on collagen I, IV, and VI-coated plates for cell adhesion assays and inflammatory SMCs (iSMCs) were established by culture in flexible silicone chambers subjected to cyclic mechanical stretch. HUVECs were cultured in iSMC-conditioned medium, followed by evaluation of their viability, apoptosis, and function, and determination of VEGF (vascular endothelial growth factor) -A and integrin levels by western blotting. Aneurysm tissue contained fewer SMCs and lacked ECs. In aneurysm walls, more matrix metalloproteinase (MMP) -1, MMP-3, and apoptotic cells were detected, accompanied by decreased collagen IV and VI levels. Cell adhesion assays revealed that more HUVECs were attached in collagen IV and VI-coated plates compared with controls. iSMC-conditioned medium significantly reduced HUVEC viability and apoptosis showed an increased trend; however, the difference was not significant. iSMC medium also reduced tube formation and migration of HUVECs. Moreover, iSMC medium reduced HUVEC expression of VEGF-A, integrin α1, integrin α2, and integrin β. Our data demonstrate a lack of SMCs and ECs in aneurysm walls, accompanied by elevated MMP and decreased collagen levels. In vitro assays showed that iSMCs induced reduction in EC adhesion, and caused EC dysfunction. Understanding of the relationships among SMC, EC, and collagens during aneurysm progression provides an additional therapeutic option for prevention of cerebral aneurysm progression.
Introduction
Cerebral aneurysms are the main cause of subarachnoid hemorrhage (SAH) and occur in 1% to 2% of the population 1 . Although the 1- and 5-year risks of aneurysm rupture are only 1.4% and 3.4% 2 , respectively, the morbidity and mortality associated with cerebral aneurysm development and rupture are not insignificant, as aneurysmal SAH is associated with approximately 30%–40% mortality 3,4 , despite modern neurosurgical intensive care. Moreover, almost half of survivors are left disabled. Hence, prevention of cerebral aneurysm progression and rupture is a clinical imperative.
The detailed natural history of cerebral aneurysms is incompletely understood. Although many researchers have investigated the pathological changes in smooth muscle cells (SMCs), endothelial cells (ECs), and the extracellular matrix (ECM), studies focusing on interactions between inflammatory SMCs (iSMCs) and ECs are insufficient.
We hypothesized that iSMCs can induce collagen degradation and EC dysfunction, which negatively impact cerebral aneurysm progression. In this study, we explored the pathological changes during cerebral aneurysm development and the mechanisms underlying EC changes exposed to iSMC-conditioned medium in vitro.
Materials and Methods
Ethics Statement
The study was reviewed and approved by the Ethics Committee of Huashan Hospital, Fudan University. Each participant provided their written informed consent to participate in this study.
Tissue Sample Preparation and Immunohistochemistry
Five aneurysms and five superficial temporal artery (STA) samples were collected for histological analysis. Aneurysm domes were removed after microsurgery clipping and STAs were extracted from patients suffering from intracranial tumors using an extended pterional approach. Tissue samples for histology were fixed in 4% formalin. After paraffin embedding, specimens were sliced into 5-μm-thick sections using a microtome (model RM2165, Leica microsystems, Nussloch, Germany), as previously described. Sections were randomly chosen from each sample for hematoxylin-eosin (H&E) and Masson’s Trichrome staining (Sigma, St. Louis, MO, USA), or immunohistochemistry (IHC). Anti-von Willebrand Factor (vWF, Abcam, Cambridge, MA, USA), anti-alpha smooth muscle actin (αSMA, Dako, Carpinteria, CA, USA), anti-Angiotensin II Type 1 Receptor (AT1, Novus Biologicals, Cambridge, UK), anti-MMP (matrix metalloproteinase)-1 (Millipore, Billerica, MA,USA), anti-MMP-3 (Millipore), anti-collagen IV (Abcam), and anti-collagen VI (Abcam) primary antibodies were used for IHC. 3,3′-diaminobenzidine (DAB) plus chromogen (Thermo Fisher Scientific, Waltham, MA, USA) was used for substrate visualization, according to the manufacturer’s protocol. An ApopTag® Peroxidase In Situ Apoptosis Detection Kit (Millipore, Merck KGaA, Darmstadt, Germany) was used for the terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) assay, according to the manufacturer’s protocol.
Cell Adhesion Assay
For cell adhesion assays, 24-well plates coated with collagen I (Sigma, St. Louis, MO, USA), collagen IV (RayBiotech, Norcross, GA, USA), and collagen VI (RayBiotech) were used. For the Con group, plates were coated with collagen I; COL IV group plates were coated with collagens I and IV; COL VI group plates were coated with collagens I and VI; and COL IV/VI group plates were coated with collagens I, IV, and VI. Human umbilical vein endothelial cells (HUVECs; 5 × 104) were transferred to 24-well plates and set on an orbital shaker agitating at 50 rpm. Medium was removed after 3 or 6 h. Calcein acetoxymethyl (calcein AM, AAT Bioquest, Sunnyvale, CA, USA) was added to detect live attached HUVECs. Cells were examined by fluorescence microscopy (Carl Zeiss, Oberkochen, Germany). Numbers of cells were calculated in each high field (HF).
Inflammatory Smooth Muscle Cell Culture
Human brain vascular smooth muscle cells (HBVSMCs) were obtained from ScienCell Research Laboratories (Carlsbad, CA, USA) and cultured in smooth muscle cell medium (SMCM, ScienCell Research Laboratories) at 37°C in a 5% CO2 incubator. The medium was changed every 3 days and collected for use for treatment of HUVECs (SMC-NC group). HBVSMCs (5 × 104) were transferred into six-well flexible silicone rubber BioFlexTM plates (Flexcell International Corporation, Hillsborough, NC, USA) and cultured at 37°C in a 5% CO2 incubator for 48 h. Stretch plates were set in an FX-5000TM Flexcell Tension PlusTM unit (Flexcell International Corporation), which elongated the cells by 15% at 1 Hz. After 72 h of cyclic mechanical stretching, medium was collected and used to treat HUVECs (iSMC-NC group).
HUVEC Culture and Viability Assay
HUVECs were cultured in DMEM (Gibco, Carlsbad, CA, USA) supplemented with 10% fetal bovine serum (FBS; Gibco) and 1% penicillin-streptomycin at 37°C, in a 5% CO2 humidified atmosphere. Medium was changed every 3 days. HUVECs were transferred in 96-well plates (1 × 103 cells per well). Cells treated with SMCM for 24 h were the control group (NC). HUVECs treated with SMC or iSMC-conditioned medium for 24 h were the SMC-NC and iSMC-NC groups, respectively. After treatment, medium was removed and HUVECs cultured in DMEM. Cell proliferation rates were estimated using CCK-8 assays (CCK-8; Dojindo Molecular Technologies, Japan) after 24, 48, and 72 h. Absorbance was detected using a TECAN infinite M200 plate reader (Tecan Trading AG, Switzerland) at 450 nm. After 24 h, viable cells were stained using calcein AM and enumerated for statistical analysis.
Apoptosis Assay
Apoptosis rates of HUVECs (2 × 104) from the SMCM, SMC-NC, and iSMC-NC groups were evaluated using an Alexa Fluor® 488 Annexin V/Dead Cell Apoptosis Kit (Thermo Fisher Scientific). Cells were detached with trypsin, centrifuged at 1000 × g for 5 min and then resuspended in 100 μl of binding buffer containing 5 μl of Annexin V. Then, 1 μl of propidium iodide was added and mixed, followed by a 15-min incubation in the dark. Subsequently, 400 μl of binding buffer was added to resuspend the cells, which were analyzed using a Gallios Flow Cytometer (Beckman Coulter, Brea, CA, USA). Experiments were repeated three times.
Tube Formation, Transwell, and Scratch Tests
The individual wells of a 96-well plate were coated with 30 μl Matrigel basement membrane matrix (BD Biosciences, Franklin Lakes, NJ, USA), and the plate placed into a 37°C cell culture incubator for 45 min. Next, 3 × 105 cells from the NC, SMC-NC, and iSMC groups were gently added to each gel-coated well. After 20 h, cells were examined. Images were acquired and analyzed for tube formation using the Wimasis image analysis software program. Covered area and total tube length parameters were used to estimate the level of tube formation.
Transwell assays were also used to assess changes in cell viability in 8-μm-pore transwell chambers (Costar, Corning, NY, USA). The upper side of insert membranes was coated with Matrigel (BD Biosciences). Cells (1 × 104) were pre-treated with SMCM, SMC-NC, and iSMC-NC for 24 h then transferred into the upper chambers in 200 µl of serum-free medium, and 600 µl of medium containing 5% FBS was added into the lower chamber as the chemoattractant. After 12 h incubation, the cells on the upper surface of the insert membrane were removed using a cotton swab. Cells on the lower surface of the insert membrane were fixed in methanol for 20 min and then stained with 1% crystal violet for 30 min. Numbers of cells on lower surfaces of membranes were counted under a microscope and then photographed. All experiments were performed in triplicate.
For the scratch assay, HUVECs were placed into 24-well plates and incubated for 48 h. Then, a pipette tip was used to scratch the cell monolayer across the center of each well. Detached cells were removed by washing, and the remaining adherent HUVECs treated with SMCM, SMC-NC, or iSMC-NC for 24 h. Cells were then fixed with 4% paraformaldehyde for 10 min and images acquired.
Western Blotting
HUVECs were treated with SMCM, SMC-NC, or iSMC-NC for 24 h, then total proteins extracted using RIPA buffer containing phenylmethanesulfonylfluoride (PMSF). After a 30-min incubation on ice, protein lysates were centrifuged at 13,000 rpm for 15 min and supernatants collected. Protein concentrations were measured using the BCA Protein Assay Kit (Pierce, Rockford, IL, USA). Ten micrograms of each protein sample were subjected to 8% SDS-PAGE and transferred to PVDF membranes, which were then blocked with 5% BSA in TBS containing 0.1% Tween-20 at room temperature for 1 h and incubated with primary antibodies diluted in blocking buffer at 4°C overnight. After washing with PBS containing 0.1% Tween, secondary antibodies were added for incubation at room temperature for 1 h. Western blot images were captured using a LI-COR Odyssey Scanner. Primary antibodies to detect human VEGF-A (Abcam), integrin α1 (Abcam), integrin α2 (Abcam), integrin β1 (Abcam), and GAPDH (Cell Signaling Technology, Danvers, MA, USA) were used.
Statistical Analysis
Statistical analyses were performed using IBM SPSS Statistics version 19 (SPSS Inc., Chicago, IL, USA), and graphs generated in Graph-Pad Prism version 5.01(GraphPad Software Inc., La Jolla, CA, USA). Images from the tube formation and migration assays were acquired and analyzed using the Wimasis image analysis software program (Wimasis GmbH, München, Germany). Two-way ANOVA tests followed by the Bonferroni tests were used for comparisons of the number of cells in cell adhesion assays, absorbance OD for CCK-8 analysis, numbers of cells for live cell staining, percentages determined by apoptosis assay, covered area and total tube length in tube formation assays, number of cells in transwell assays, and scratch area in migration assays. p values < 0.05 were considered significant.
Results
Histological Assessment and IHC
HE- and Masson’s trichrome-stained sections were examined under low magnification. STA sections showed obvious vascular structures and a more integrated SMC arrangement, while in aneurysm sections SMCs were sparser and cell loss and asymmetrical collagen distribution were observed. Fewer vWF+ cells were detected via IHC in the aneurysm intima compared with those in STA samples. In addition, fewer αSMA+ and AT1+ cells and more MMP-1+ and MMP-3+ cells were detected in aneurysm tunica media. Moreover, fewer collagen type IV+ and VI+ cells were visible in aneurysm specimens. In STA samples, the majority of collagen was located around SMCs, whereas in aneurysm tissue, type IV and VI collagens were rarely detected. By TUNEL assay, more apoptotic cells were observed in aneurysm tissue samples (Fig. 1).

HE staining, Masson’s trichrome staining, immunohistochemistry and TUNEL assay. Photomicrographs showing the histological features of STA and aneurysm tissue samples. Representative photomicrographs showing HE-stained and Masson’s trichrome-stained vascular tissues. Immunohistochemical detection of vWF, αSMA, AT1, MMP-1, MMP-3, collagen IV, and collagen VI and evaluation of changes in cell numbers and cytokine expression. TUNEL assay; stained nuclei indicate apoptosis. Scale bar = 200 μm.
Cell Adhesion Assay
After 3 h shaking, significantly more live cells were observed in cells cultured in plates coated with collagens I, IV, and VI (COL IV/VI group) compared with control plates coated with collagen I alone (Con group). There were also elevated numbers of cells on plates coated with collagens I and IV (COL IV group) and with collagens I and VI (COL VI group); however, the increase was only significant for the COL VI group (Fig. 2A). After 6 h, numbers of cells in all experimental groups (COL IV, COL VI, and COL IV/VI) were significantly increased compared with the Con group (Fig. 2B).
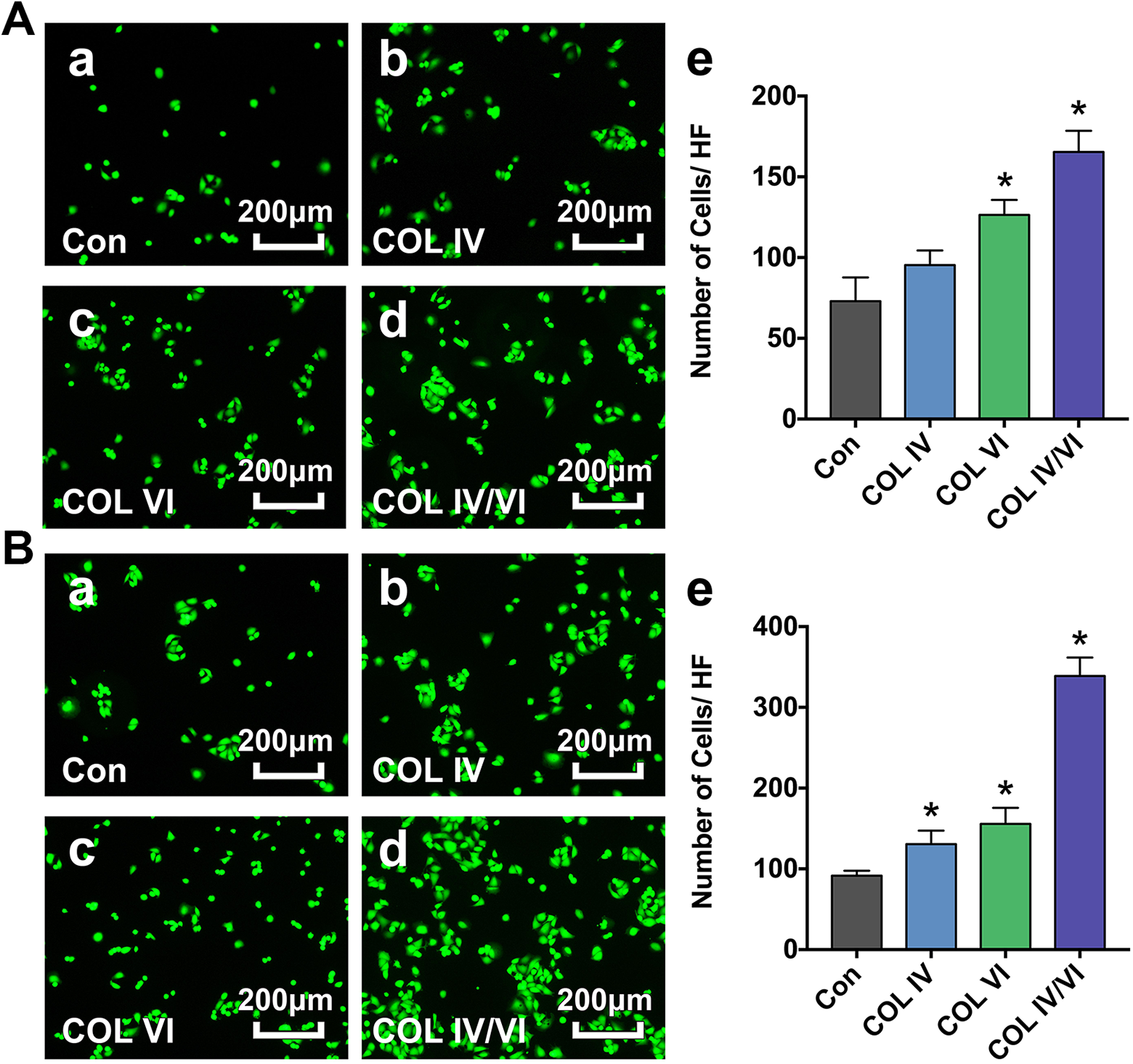
Cell adhesion assay. A: Representative photographs showing attached viable HUVECs in plates coated with collagen I alone (Con) (a), collagens I and IV (COL IV) (b), collagens I and VI (COL VI) (c), and collagens I, IV, and VI (COL IV/VI) (d) groups after orbital shaking for 3 h. Bar graph (e) demonstrates the number of cells per high field (HF). Bar = 200 µm. Data are presented as mean ± SD, n = 5 per group, *p < 0.05 for COL IV, COL VI, or COL IV/VI groups versus the Con group. B: Representative photographs showing attached viable HUVECs in the Con (a), COL IV (b), COL VI (c), and COL IV/VI (d) groups following orbital shaking for 6 h. Bar graph (e) demonstrates the number of cells per high field (HF). Bar = 200 µm. Data are presented as mean ± SD, n = 5 per group, *p < 0.05 for COL IV, COL VI, or COL IV/VI groups versus the Con group.
Viability and Apoptosis Assays of Attached Cells
Cell viability assays using the CCK8 kit indicated that, compared with HUVECs treated with unconditioned SMC medium (NC group) and those treated with SMC-conditioned medium (SMC-NC group), there were significant decreases in viability of HUVECs after 24, 48, and 72 h exposure to iSMC-conditioned medium (iSMC-NC group). This indicates that iSMCs can reduce HUVEC viability; the difference was most marked at 72 h (Fig. 3A). Consistently, immunofluorescence images acquired at 72 h showed the same trend, with a significant decrease in cells observed in iSMC-NC group compared with the NC and SMC-NC groups (Fig. 3B). Although iSMCs promoted HUVEC apoptosis, the percentage of apoptotic cells was not significantly different among experimental groups (Fig. 3C).
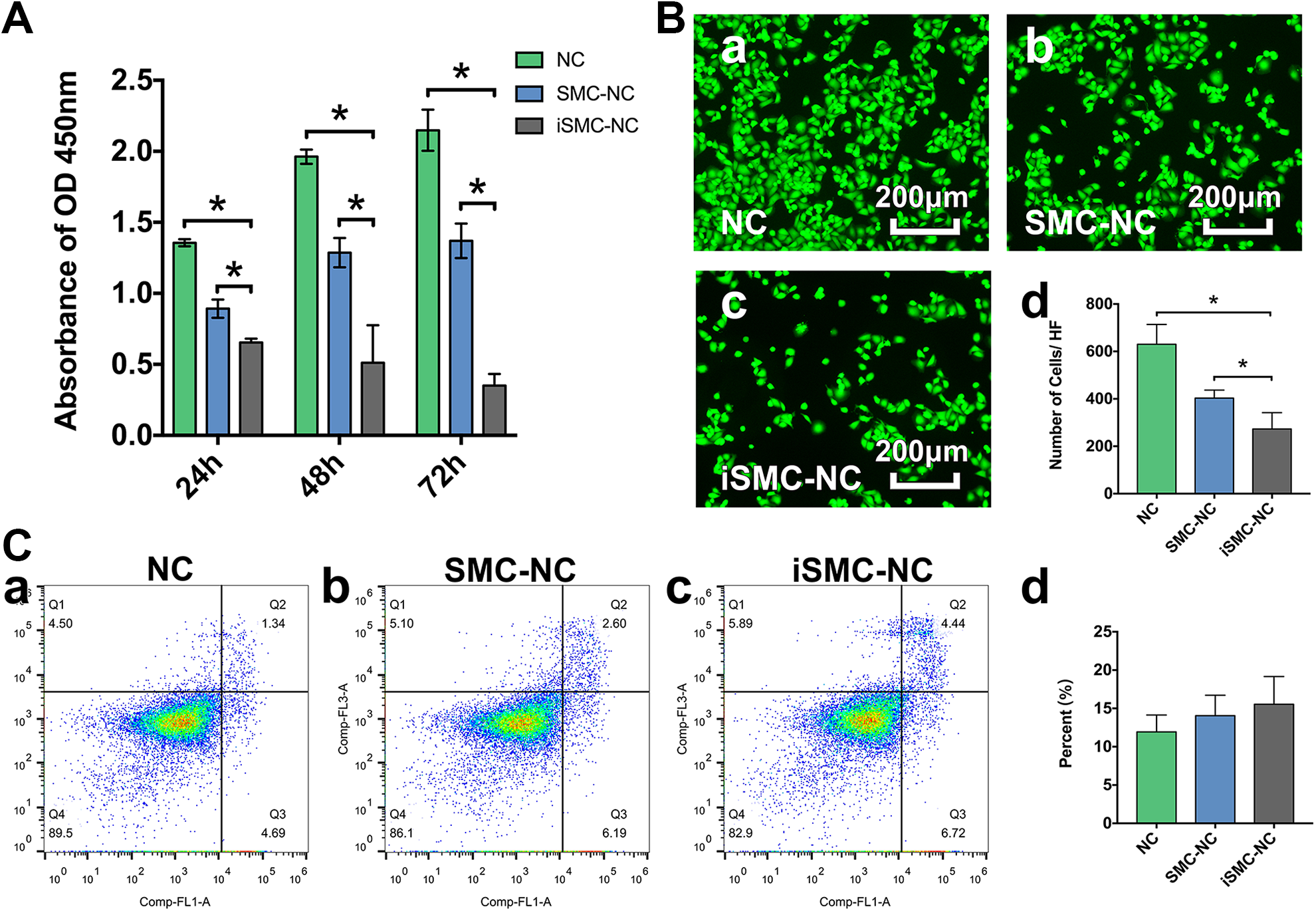
Cell viability and apoptosis. A: Bar graph showing changes in treated HUVEC viability after 24, 48, and 72 h. Data are presented as mean ± SD, n = 4 per group, *p < 0.05 for cells cultured in unconditioned SMC medium (NC) or SMC-conditioned medium (SMC-NC) versus those grown in iSMC-conditioned medium (iSMC-NC). B: Representative photographs showing viable HUVECs in the NC (a), SMC-NC (b) and iSMC-NC (c) groups after treatment for 72 h. Bar graph (d) illustrating the number of cells per HF. Bar = 200 µm. Data are presented as mean ± SD, n = 4 per group, *p < 0.05 for NC or SMC-NC groups versus the iSMC-NC group. C: Detection HUVEC apoptosis after treatment for 24 h. Representative flow cytometry images of (a) HUVECs and (b) cells treated with SMC- or (c) iSMC-conditioned medium for 24 h. Bar graph shows the percentage of apoptotic cells. Data are expressed as the mean ± SD, n = 4 per group, *p < 0.05 for the NC or SMC-NC groups versus the iSMC-NC group.
iSMC-Conditioned Medium Induced Functional Changes in HUVECs
Tube formation assays demonstrated that treatment with iSMC medium for 24 h reduced HUVEC tube formation compared with the NC and SMC-NC groups (Fig. 4A). In transwell invasion assays, iSMC medium treatment for 72 h reduced the invasive capacity of HUVECs (Fig. 4B). Furthermore, we performed a scratch assay to assess the degree of HUVEC migration and found that treatment with iSMC medium for 24 h significantly decreased cell migration (Fig. 4C).

Tube formation, transwell assay and cell migration assays. A: Representative photographs showing tube formation in HUVECs grown in unconditioned SMC medium (NC) (a), SMC-conditioned medium (SMC-NC) (b) and iSMC-conditioned medium (iSMC-NC) (c). Bar graphs illustrating the covered area (d) and total tube length (e). Bar = 200 µm. Data are presented as mean ± SD, n = 3 per group, *p < 0.05 for NC or SMC-NC groups versus the iSMC-NC group. B: Representative images of invasive cells in the lower chamber stained with crystal violet. Results of transwell assays to evaluate the migration ability of HUVECs in the NC (a), SMC-NC (b) and iSMC-NC (c) groups. Bar graph (d) illustrates the number of cells per HF. Bar = 200 µm. Data are presented as mean ± SD, n = 4 per group, *p < 0.05 for the NC or SMC-NC groups versus the iSMC-NC group. C: Representative images of HUVEC migration in the NC (a, b), SMC-NC (c, d), and iSMC-NC (e, f) groups in the scratch assay. Bar graph (g) illustrates the percentage of the scratched area covered. Bar = 200 µm. Data are presented as mean ± SD, n = 3 per group, *p < 0.05 for the NC or SMC-NC groups versus the iSMC-NC group.
iSMC-Conditioned Medium Reduced HUVEC VEGF-A and Integrin Expression
Next, we explored changes in VEGF-A, integrin α1, integrin α2, and integrin β1 expression in HUVECs subjected to iSMC-conditioned medium by western blotting. The results indicated decreased VEGF-A, integrin α1, integrin α2, and integrin β1 expression compared with the NC and SMC-NC groups (Fig. 5).

Changes in VEGF-A and integrin levels in HUVECs subjected to iSMC-conditioned medium. Western blotting analysis of VEGF-A, integrin α1, integrin α2, and integrin β1 in HUVECs grown in unconditioned SMC medium (NC), SMC-conditioned medium (SMC-NC), and iSMC-conditioned medium (iSMC-NC). GAPDH was used as a loading control. Representative images of experiments repeated three times are presented.
Discussion
Cerebral aneurysm is a disease of brain arteries manifesting as abnormal enlargement of the vessel lumen. The mechanisms underlying the genesis and progression of cerebral aneurysms remain unknown. Given the high rates of disability and lethality associated with SAH caused by aneurysm rupture, elucidation of the biochemical mechanisms underlying this condition would be valuable to assist in prediction of aneurysm progression and rupture.
Vascular SMCs, ECs, and ECM maintain physiological function in healthy vessels, and pathological vascular wall instability occurs in response to physical or chemical stimulation. Our histological observations showed that there were fewer SMCs in aneurysm walls, along with decreased numbers of αSMC+ and AT1+ cells. Conversely, more MMP-1 and MMP-3-positive cells were observed. Dynamic changes and eventual loss of the media layer contribute to aneurysm formation and rupture. Previous histological studies have demonstrated that normally contractile SMCs respond to environmental cues by undergoing phenotypic changes, causing them to manifest a pro-inflammatory, pro-remodeling, and dedifferentiated phenotype 5 –8 . The pro-inflammatory phenotype is characterized by reduced levels of the contractile elements of SMCs, accompanied by increased levels of transcription factors involved in promoting inflammation, recruitment of reactive oxygen species, and matrix remodeling 9 –11 . This SMC phenotype switch has been identified as potentially promoting aneurysm progression 12 –18 .
Degeneration of the ECM is primarily induced by secretion of inflammatory cytokines and cell infiltration 19 –21 . Our previous comparative proteomic analysis demonstrated that collagen IV and VI levels were reduced in comparisons of aneurysm and STA tissues 22 . In this study, we observed that levels of collagen IV and VI were clearly decreased in the aneurysm wall by IHC. Collagen IV is distributed around SMCs and depends on the population and density of SMCs. Collagen VI contributes to strengthening the aneurysm wall against hemodynamic stress, and reduced levels of this protein may be associated with aneurysm growth. In our cell adhesion assays, wells lacking collagen IV and VI coating contained fewer attached ECs. Hence, decreased collagen IV and VI in the aneurysm wall likely leads to increased opportunities for EC escape. Previous studies have proven that in situ collagen can be used for aneurysm growth risk assessment, and radiocarbon birth dating of collagen has been used as an indicator of molecular changes 23,24 .
ECs have various important functions, including maintenance of vascular hemostasis and secretion of several vasoactive and anti-thrombogenic mediators. In our former research, we established iSMCs using cyclic mechanical stretching. When treated with iSMC-conditioned medium, the viability and function of HUVECs were generally decreased. Recent evidence points to damage of the vascular endothelium as the initiating event, leading to the establishment, inflammatory cascade production, progression, and rupture of intracranial aneurysms 25 –27 .
Integrins have a crucial role in HUVEC adhesion, with integrin α1, α2, and β1 vital to interactions between ECs and collagens. Cells are mechanically integrated structures, in which the ECM and actin cytoskeleton are connected by integrins and focal adhesion proteins. iSMC-induced integrins are down-regulated in ECs, and ECM remodeling leads to EC escape from the aneurysm wall. Hemodynamic forces exerted by flowing blood on vessel walls lacking ECs induce vascular remodeling and lead to aneurysm formation, growth, and rupture 28 –30 .
VEGF is key in cerebral aneurysm progression 31,32 . VEGF-A is the most potent pro-angiogenic protein described to date. It induces proliferation, sprouting, and tube formation of ECs 33 , and is also a potent EC survival factor that can induce the expression of anti-apoptotic proteins in ECs 34,35 . Our data indicate that treatment of HUVECs with iSMC-conditioned medium reduces their expression of VEGF-A, suggesting that this may contribute to EC dysfunction.
Under physiological conditions, aneurysm stability depends not only on the balance between cell proliferation and dysfunction, but also on the equilibrium between the regeneration and degradation of the ECM 36 –38 . Taken together, our data indicate that iSMCs induce collagen degradation and EC dysfunction, which could accelerate cerebral aneurysm progression (Fig. 6).

A schematic model illustrating the mechanism underlying iSMC-induced EC changes in cerebral aneurysms A: As SMCs switch to an inflammatory phenotype, cell contraction ability decreases and higher levels of MMPs are secreted. iSMCs induce collagen degeneration, which induces EC escape. In addition, iSMCs induce EC dysfunction. EC escape and dysfunction expose the vascular tunica media to blood flow, which eventually attenuates inflammation and slows aneurysm progression.
This study has several limitations. First, although iSMCs act on HUVECs via regulation of integrins and VEGF-A, other pathways, such as the Akt and Nrf-2 pathways, could also contribute to these processes. Second, we have yet to determine how iSMCs act on ECs and the underlying mechanism should be explored further in future. Further in vivo studies will provide more evidence to evaluate the changes that occur during the process of aneurysm growth.
Footnotes
Acknowledgements
Thanks are due to Bo Yang for diagram drawing and Yingjun Guo for manuscript checking.
Author Contributions
Peixi Liu: Conception and design of study, collection and/or assembly of data, data analysis and interpretation, manuscript writing, and final approval of manuscript. Yuan Shi: Database input and data interpretation, manuscript writing, and final approval of manuscript. Zhiyuan Fan: Data collection and analysis. Yingjie Zhou: In vitro assay data analysis, histological examination. Yaying Song: Data analysis and interpretation, manuscript writing. Database Input and Data Interpretation. Yingjun Liu: Sample analysis. Qingzhu An: Provision of study and revision and final approval of manuscript. Wei Zhu: conception and design of study and revision and final approval of manuscript. Peixi Liu and Yuan Shi contributed to this work equally. Qingzhu An and Wei Zhu are co-corresponding authors of this article.
Ethics Approval
Our study was approved by Ethics Committee of Huashan Hospital, Fudan University (Reference number: 2017-263).
Statement of Human and Animal Rights
All procedures in this study were conducted in accordance with Ethics Committee of Huashan Hospital, Fudan University (Reference number: 2017-263). This article does not contain any studies with animal subjects.
Statement of Informed Consent
All patients whose samples were used in the study had signed the written consents of inform.
Declaration of Conflicting Interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This project was sponsored by the National Natural Science Foundation of China (No. 81571102 to WZ); Outstanding Academic Leaders Program of Shanghai Municipal Commission of Health and Family Planning (No. 2017BR006 to WZ); The Program for Shanghai Sailing Program, project No. 16YF1401200 (LPX). The funding organization had no role in the design or conduct of this research.