
Abstract
Hematopoietic stem and progenitor cells reside within the bone marrow (BM) microenvironment. By a well-balanced interplay between self-renewal and differentiation, they ensure a lifelong supply of mature blood cells. Physiologically, multiple different cell types contribute to the regulation of stem and progenitor cells in the BM microenvironment by cell-extrinsic and cell-intrinsic mechanisms. During the last decades, mesenchymal stromal cells (MSCs) have been identified as one of the main cellular components of the BM microenvironment holding an indispensable role for normal hematopoiesis. During aging, MSCs diminish their functional and regenerative capacities and in some cases encounter replicative senescence, promoting inflammation and cancer progression. It is now evident that alterations in specific stromal cells that comprise the BM microenvironment can contribute to hematologic malignancies, and there is growing interest regarding the contribution of MSCs to the pathogenesis of myelodysplastic syndromes (MDSs), a clonal hematological disorder, occurring mostly in the elderly, characterized by ineffective hematopoiesis and increased tendency to acute myeloid leukemia evolution. The pathogenesis of MDS has been associated with specific genetic and epigenetic events occurring both in hematopoietic stem cells (HSCs) and in the whole BM microenvironment with an aberrant cross talk between hematopoietic elements and stromal compartment. This review highlights the role of MSCs in MDS showing functional and molecular alterations such as altered cell-cycle regulation with impaired proliferative potential, dysregulated cytokine secretion, and an abnormal gene expression profile. Here, the current knowledge of impaired functional properties of both aged MSCs and MSCs in MDS have been described with a special focus on inflammation and senescence induced changes in the BM microenvironment. Furthermore, a better understanding of aberrant BM microenvironment could improve future potential therapies.
Introduction
In the bone marrow (BM), the hematopoietic stem cell (HSC) subset is involved in the production of mature blood cells throughout an individual’s lifetime 1 . A small population of HSCs reside within the BM niche and are able to self-renew and differentiate into all blood cell types supplying the required number of mature hematopoietic elements throughout an individual's life span.
HSCs are basically quiescent, but they can reversibly enter cell-cycle depending on both intrinsic transcriptional pathways and extrinsic elements such as cell-to-cell interactions and secreted factors 1 . According to the conventional hierarchical system, HSCs give rise to both lymphoid and myeloid progenitors which in turn generate all the mature immune and blood elements 2 . However, this model has been partially reconsidered, as recent studies demonstrated that the HSC population is not homogeneous and that different HSC subtypes with distinct lineage differentiation potential can be identified 1 .
More in detail, HSC may present increased propensity to differentiate toward myeloid (myeloid-biased HSC) or lymphoid (lymphoid-biased HSC) lineage.
In healthy conditions, this system is balanced; during aging, its equilibrium is lost and myelopoiesis improves to the detriment of lymphopoiesis leading to the decline in the adaptive immune system with increased strain on the innate immune system. This phenomenon leads to augmented levels of basal inflammation modifying the entire BM microenvironment which normally regulates the maintenance of HSC quiescence and long-term reconstitution capability 1 .
Therefore, when age-related changes occur, HSCs are exposed to an aberrant pattern of extrinsic factors such as oxygen concentration, cytokines, and hormones that can induce genetic and epigenetic alterations increasing the tendency of hematological malignanciy insurgence 3,4 .
Studies performed in animal models showed that old HSCs are located farther from the endosteum with respect to their younger counterpart and that they are subjected to a higher oxygen concentration that causes DNA damage which increases the mutational rate 4 . Furthermore, old HSCs have been demonstrated to be less efficient and to have reduced homing and engraftment capacity. In a microenvironment where the efficiency of resident cells is compromised and their overall fitness is reduced, the probability to select a clone harboring an adaptive oncogenic mutation is increased 4 .
It is well known that the accumulation of somatic DNA mutations is a hallmark of aging, particularly in proliferating tissues, which over time become a mosaic of cells with different genotypes due to the clonal expansion of single de novo mutations 5 . Recent human studies have shown that normal aging is associated with an increased frequency of somatic mutations in the hematopoietic system, which provide a competitive growth advantage to the mutant cell and allow its progressive clonal expansion (clonal hematopoiesis) 6 –10 . This acquired clonal mosaicism in the hematopoietic system of healthy individuals predicts an increased risk of subsequent hematological malignancies 6 –8 , but it has also been associated with a higher prevalence of vascular complications of diabetes, greater incidence of atherosclerotic conditions, and increased frequency of cardiovascular disease-related death 11 .
According to this hypothesis, the incidence of myeloid hematological malignancies is increased in the elderly and seems to be related to the accumulation of mutations in replicating HSCs 12 .
Myelodysplastic syndromes (MDSs) are a heterogeneous group of age-related hematological malignancies affecting BM HSCs 13 , characterized by aberrant hematopoiesis, peripheral blood cytopenia, and a tendency for acute myeloid leukemia (AML) evolution.
In early-stage MDS, enhanced apoptosis, increased phagocytosis, and reduced differentiation of HSCs result in peripheral blood cytopenia. During the progression of the disease, accumulation of mutations in HSCs leads to a differentiative arrest and to an enhanced proliferation of clonal cells with reduced apoptosis rates adn a tendency for high-risk/AML evolution 14 .
Although the most important event in MDS pathology appears to be a molecular defect in HSCs, evidence suggests that ineffective hematopoiesis may also result from abnormalities in the BM microenvironment, including hematopoietic–stromal interactions with deregulated production of secreted factors and altered immune regulation 15 .
The BM niches are defined as cellular and molecular microenvironments that cooperate with cell-intrinsic mech anisms to maintain and regulate stem cell functions 16 . The complexity of the niche is attributed to the fact that it simultaneously contains stem cells, progenitor cells, and terminally-differentiated cells, such as HSCs, precursors for osteoclasts, responsible for bone reabsorption, and mesenchymal stromal cells (MSCs) that progressively differentiate to give rise to adipocytes or to mature osteoblasts producing the BM matrix 16 . Functional and dynamic relationships among different cells found in the BM allow autocrine, paracrine, and endocrine activities via locally produced soluble factors, controlling the activity of the BM microenvironment 17,18 .
Recently, BM-MSC biology in MDS has been well investigated, and data obtained from both in vivo and in vitro models reported altered molecular and functional features of these cells, such as altered cell-cycle regulation with impaired proliferative potential, abnormal cytokine secretion, and a dysregulated gene expression profile 19 –21 . Moreover, data from in vivo murine models suggested that the pathogenesis of MDS involves BM-MSCs demonstrating the presence of an aberrant cross talk between hematopoietic and stromal compartments that could be responsible for ineffective hematopoiesis 22,23 . This active relationship between HSC and BM microenvironment could be controlled by several bioactive factors secreted from the stromal cellular compartment, involving immunomodulatory effects, maintenance of stemness activity, apoptosis, and senescence regulation 24 –29 . However, little is known about the aged MSC secretome and its potential autocrine/paracrine role.
In this review, typical senescence-associated markers (SAMs) and impaired functional properties of aged MSCs have been described, focusing on altered microenvironments in aging-related pathological conditions, such as MDS. Here, the changes that occur in the BM microenvironment and in MSCs will be discussed in more detail, and how these changes affect HSC fate will be analyzed. A detailed understanding of these mechanisms may help to define novel targets for diagnosis and possibly therapy.
MSCs and Aging-associated Alterations
Human MSCs/stem cells are nonhematopoietic cells capable of self-renewal and multilineage differentiation into various tissues of mesodermal origin. Located in the hematopoietic niche, BM-MSCs represent together with osteoblasts an essential population that provides to maintain HSC homeostasis and contributes to support hematopoiesis. In particular, MSC and osteoblasts express a number of adhesion molecules and secreted factors that regulate blood regeneration throughout life by contributing to HSC and progenitor cell maintenance, self-renewal, and differentiation 15 .
Aged MSCs generally perform less well than their younger healthy counterparts, 30 and growing evidence strongly suggests that cellular senescence contributes to aging and age-related diseases. Senescence is a persistent cell-cycle arrest that occurs in response to stressful signals in cells previously able to replicate in order to prevent cancer insurgence and progression 31 .
Mitotically competent cells respond to many stressors adopting a state of permanent cell-cycle arrest by cellular senescence. These stressors can be represented by oxidative stress 32,33 , chemotherapeutic agents 34 –36 , genotoxic stresses/DNA damage, perturbations to chromatin organization, and strong mitogenic signals. Senescence cells undergo irreversible growth arrest but continue to be metabolically active. They develop a large, flat morphology showing a senescence-associated β-galactosidase activity 30 . Additionally, they show changes in gene expression profile, dysfunctional telomeres, persistent DNA damages, and a distinct heterochromatin structure, indicating a specific senescence-associated phenotype 37 .
Moreover, senescent cells secrete factors that affect vital and tightly regulated processes, such as cell growth and migration, tissue architecture, blood vessel formation, and differentiation and their inappropriate presence can disrupt tissue structure and function.
Among these secreted factors, there are high levels of several potent inflammatory cytokines able to affect the behaviors of neighboring cells 38 , involving chronic inflammation in the initiation or support of several age-related diseases. In particular, the strong oxidants produced by some immune cells can alter and remodel the tissue environment, promoting cell/tissue dysfunction and stem-cell niche impairment (Fig. 1). Oxidative damage and the general inflammatory environment can initiate carcinogenesis and promote cancer by suppressing immune surveillance and stimulating malignant phenotypes 39 –41 .
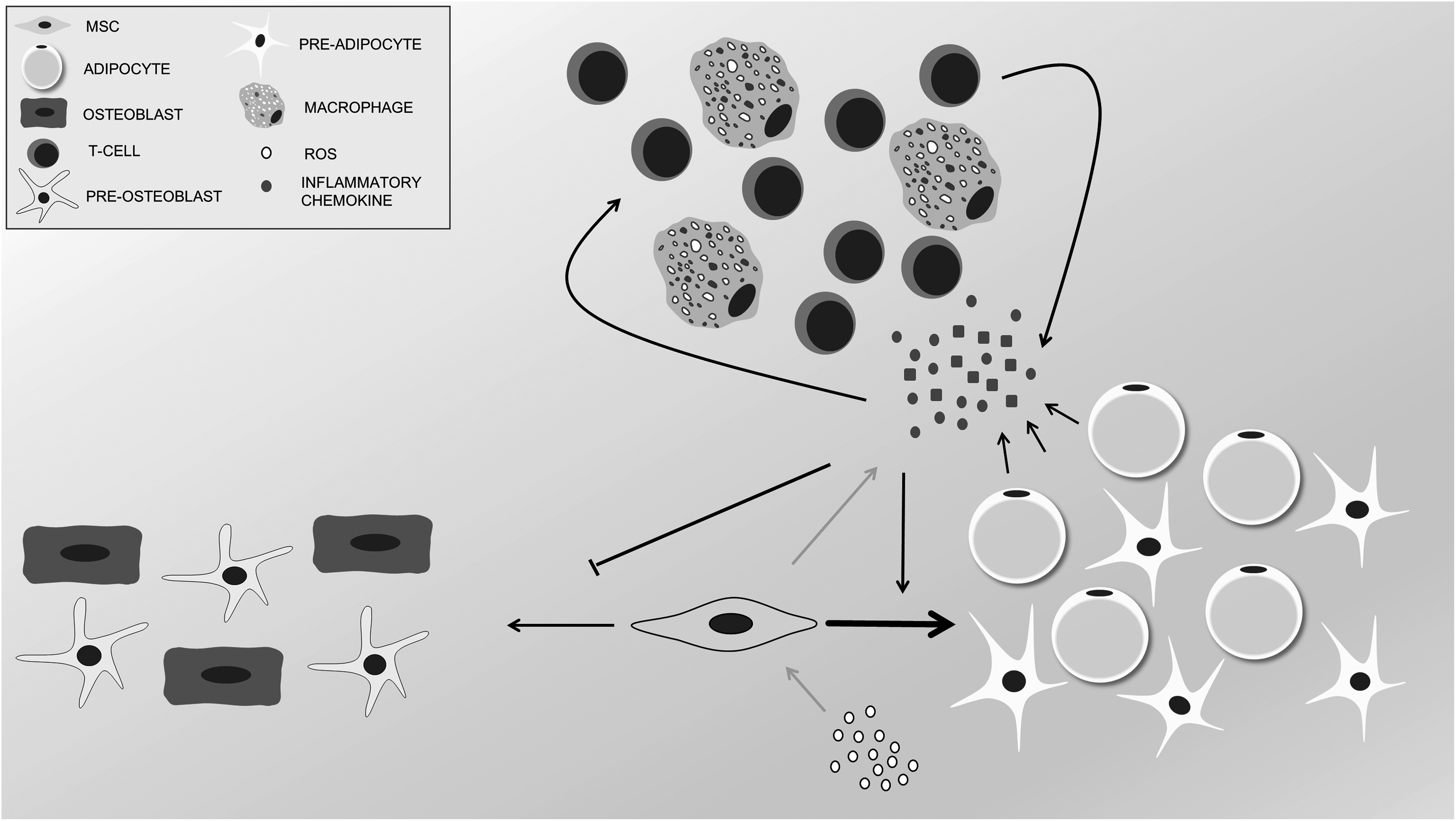
Aged bone marrow microenvironment. Oxidative stress and the increment of reactive oxygen species during aging stimulate the secretion of inflammatory cytokines by mesenchymal stromal cells (MSCs) through Nuclear factor Kappa-light-chain-enhancer of activated B cells (NFkB) pathway. Increased levels of inflammatory cytokines lead to infiltration of immune cells like T cells and macrophages. Altogether, these changes in the composition of bone marrow microenvironment result in an osteoblast-to-adipocyte shift in MSC differentiation potential which through secreted inflammatory cytokines and adipokines keep maintaining this inflammatory circle.
The molecular mechanisms that characterize senescence are still poorly understood. Two fundamental mechanisms have been hypothesized to describe the ways this process may be regulated: replicative senescence might either be the result of a specific program driven by genes or rather be evoked by stochastic events 42,43 .
Analysis of the gene expression profile identified more than 5,000 genes, including 31 microRNA (miRNA), differentially expressed in senescent MSCs compared to proliferating cells. These genes affect several cellular functions, including cell growth and proliferation, cell cycle, cell death, and cellular movement. Senescent cells showed an enrichment of upregulated genes in pathways which regulate the cellular morphology, while genes involved in the cell-cycle activities were mostly downregulated. These categories are perfectly in line with the reduced proliferation potential and changes in cellular cytoskeleton observed in senescent cells 43 . These data strengthen the hypothesis that senescence follows a fixed program where genes involved in the proliferation machinery are downregulated. On the other hand, these findings do not rule out the possibility that the activation of a specific “senescence program” could be the consequence of accumulation of cellular defects such as oxidative stress, telomere loss, or DNA damage 43 .
The factors that might mediate this process are yet unknown but epigenetic modifications involving DNA methylation changes are clearly affected by cellular senescence 44 , in particular along promoter regions, which become either hypermethylated or hypomethylated 44 –46 . Reports evidenced that DNA methylation status changes in genes related to DNA replication, cell-cycle regulation, DNA repair, differentiation, and several metabolic processes.
For example, it has been demonstrated that the homeobox gene distal-less homeobox 5 gene (DLX5), involved in osteoblastic differentiation 47 , becomes hypermethylated upon long-term culture affecting the differentiation capability of MSCs 48,49 . Methylation changes have also been described in several tumor suppressor genes whose reduced expression may increase the tendency to malignant transformation 50 .
Some of these changes can be observed both in MSCs isolated from old patients and in cells aged in “in vitro models” and are highly reproducible, suggesting that cellular senescence represents a specific epigenetic developmental program rather than an ensemble of stochastic events and that it can occur physiologically during aging or be the consequence of environmental stressors 51 .
Many reports investigated the effect of MSC senescence on migratory ability, differentiation potential, immunomodulation ability, and tumor progression. On one hand, senescent MSCs retained an aptitude to regulate the inflammatory response on macrophages in vitro and, in part, retained their ability to inhibit lymphocyte proliferation, but on the other hand they had a severely reduced migratory capacity in response to pro-inflammatory signals 52 . Notably, many of the senescence-associated secretory phenotype components oversecreted by senescent MSCs, such as leptin, Tumor growth factor-alpha (TGF-α), Interleukin 8 (IL-8), eotaxin, Interferon gamma (IFN-γ), Vascular cell adhesion molecule 1 (VCAM1), Interferon beta (IFN-β), Interleukin 4 (IL-4), and Monocyte chemotactic protein (MCP-1), are involved in the systemic inflammatory response, decreasing the immune modulation activity of MSCs and promoting either proliferation or migration of cancer cells 52 .
Moreover, the accumulation of oxidative stress and dysregulation of key differentiation regulatory factors determine decreased differentiation potential of senescent MSCs. Age-related bone loss begins as early as 20 y in young adults, long before hormonal changes can affect bone strength and density 53 . Recent studies have shown that oxidative stress in aging mice may be an important pathogenic mechanism that leads to age-related bone loss and reduced bone strength. In addition, loss or reduced levels of sex hormones in aging mice accelerate the effects of aging on the bone by decreasing defense against oxidative stress 54 . Even though it is not clear whether oxidative stress is the main reason for age-related bone loss in humans, an increasing number of experimental and epidemiological evidence links osteoporosis to accumulated reactive oxygen species (ROSs) in the BM 55 . These ROSs not only cause tissue damage and cell senescence but also lead to BM inflammation through redox sensitive transcriptional factors such as nuclear-κ B (NF-κB) 56 –58 . NF-κB is one of the most important transcription factors that respond directly to oxidative stress conditions. After receiving an appropriate signal, NF-κB is activated and translocated to the nucleus where it stimulates the expression of IL-1, IL-6, TNF-α, and other cytokines essential to trigger an inflammatory response 59 . ROS stimulation enhances the signal transduction pathways for NF-κB activation in the cytoplasm and translocation to the nucleus 56 –58 . Under physiological condition, NF-κB activation in response to extracellular signals is short lived, and the reaction stops quickly once the signal is removed 59,60 . However, if the activation of the signal persists, such as accumulation of ROS in aged BM, the effect of NF-κB signal becomes persistent thus leading to elevated levels of pro-inflammatory cytokines in the BM. Pro-inflammatory cytokines such as TNF-α, IL-6, and MCP-1 promote a cascade of events that result in the recruitment of inflammatory T lymphocyte subsets, mast cells, monocytes, and macrophages from the blood 61 . These infiltrated immune cells in BM secrete more pro-inflammatory factors and together contribute to an inflammatory microenvironment. It is known that pro-inflammatory cytokines such as TNF-α and IL-1 also stimulate ROS generation through mitochondrial and NADH system 62 . Therefore, oxidative stress and inflammation together promote a positive feedback loop that characterizes the pathological microenvironment of aged BM 63 .
In conclusion, the knowledge of the physiological and pathological factors that influence the MSC activity is of fundamental importance to better understand the microenvironment key role in pathological conditions such as in MDS.
Moreover, aging and cellular senescence are associated with inflammation and the increased frequency of cancer progression; the analysis of the microenvironment secretome, mediating senescence, and the mechanisms that drive toward MSC growth arrest are important to develop efficient therapeutic approaches that can preserve a functional stem-cell pool and microenvironment.
Phenotypic and Functional Alterations of MSC-MDS
Although a universal senescence marker has not yet been defined, MSC-MDS in vitro display several features which suggest that these cells are encountering senescence 64 . These cells have been described as large, flat, granular, and disorganized with impaired proliferative potential and increased β-galactosidase expression 19,21,65,66 .
All these features are considered typical SAMs; therefore, many authors improved the characterization of this process, exploring more in detail the molecular and functional mechanisms behind induced cellular senescence (Table 1).
MSC-MDS alterations.a
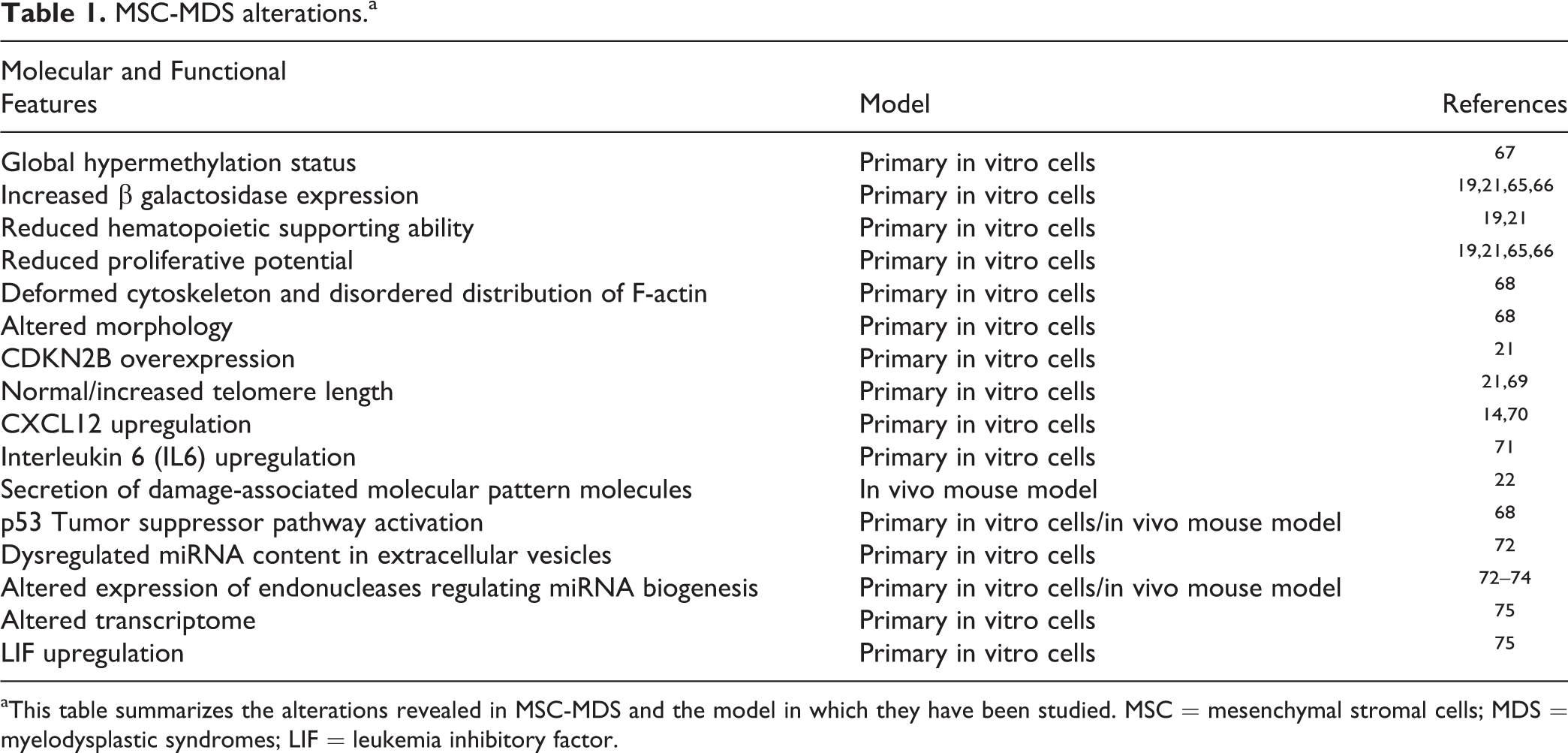
aThis table summarizes the alterations revealed in MSC-MDS and the model in which they have been studied. MSC = mesenchymal stromal cells; MDS = myelodysplastic syndromes; LIF = leukemia inhibitory factor.
The morphology of MSC-MDS has been studied through an immunofluorescence assay, which describes the actin architecture within the cells. Cells in culture displayed a deformed cytoskeleton, an increased size, numerous and longer podia, and disordered distribution of F-actin, coherently with a senescent cellular morphology68.
Beside altered morphology, MSC-MDS presented an impaired proliferative potential, and it was studied at both functional and molecular levels. In particular, Cyclin dependent kinase inhibitor 2B (CDKN2B), a cyclin-dependent kinase inhibitor, that if overexpressed leads to premature cell-cycle arrest, resulted in significantly upregulated in MSC-MDS 21 . This gene has also been evaluated HSC-MDS, demonstrating that its hypermethylation could be associated with a poor patient prognosis 76 –78 . These data suggest that HSC-MDSs are able to modify their methylation pattern to escape the CDKN2B-mediated cell-cycle arrest, evading the inhibitory signals from the BM microenvironment and taking advantage of healthy HSCs 79 .
Interestingly, in patients in complete hematological remission after azacitidine treatment, MSCs in culture recover a proliferative potential similar to donors together with a downregulation of CDKN2B 67 . A specific CDKN2B-mediated cell-cycle modulation in an age-related disease such as MDS is not surprising; indeed, this gene regulates several tumor suppressor pathways and influences key physiological processes such as replicative senescence, apoptosis, and stem-cell self-renewal 80 . The proliferative potential of cells is normally associated with telomere length, and their shortening as the consequence of recurring cellular divisions leads to a gradual loss of DNA at the end of chromosomes 81,82 . This aspect has been analyzed in MDS and it has been shown that MSC-MDS presented significantly shortened telomeres in young donors, but no significant differences were found in old donors 21 . However, some authors reported that telomere length was even increased in patients 69 . These data suggest that MSC-MDS before encountering telomere erosion caused by cumulative cellular replications is subjected to a premature cellular senescence that is presumably environmentally induced.
As part of the hematopoietic niche, MSCs in healthy conditions secrete factors that protect, renew, and maintain adult stem cells. Yet in hematological malignancies, the cytokine secretion pattern is dysregulated, and there is evidence of ongoing inflammatory processes that correlate with high concentration of TNF-α, IFN-γ, IL-1α, IL-6, IL-17, and TGF-β in peripheral blood of patients 83 –86 .
The establishment of an inflammatory microenvironment may affect resident stem cells modifying their gene expression profile and inducing genomic and epigenetic alterations involved in malignant transformation 87 –91 . TGF-β, for example, regulates cellular proliferation and differentiation and affects both early and late stages of hematopoiesis exerting suppressive effects on both erythroid and myeloid cells. TGF-β pathway is constitutively activated in CD34+ cells of MDS patients together with the activation of the downstream mediator Smad2, an important pathway inhibitor in MDS. Interestingly, it has been shown that the suppression of TGF-β signaling is sufficient to improve hematopoiesis in vitro and in vivo in a murine model of BM failure demonstrating a relevant role of this cytokine in hematological disorders 92 .
CXCL12 is another chemokine produced by MSCs and by other cells of the niche and it is involved in HSC homing and maintenance. This regulation is mediated via C-X-C chemokine receptor type 4 (CXCR4) binding, a receptor expressed on the surface of both leukemic and healthy HSCs. In pathological condition, the C-X-C motif chemokine 12 (CXCL12) expression can be higher in HSC-MDS in healthy subjects, and its interaction with CXCR4 keeps cells anchored inside the niche furnishing them protection and support 14,70 . In vitro studies also demonstrated that pediatric MSC-MDS secretes higher levels of IL-6 when compared with control 71 . IL-6 has been associated with MSC senescence and chronic inflammation; its overproduction may reduce the quiescence of HSCs and may promote their proliferation, depleting the marrow of hematopoietic progenitors. While in healthy condition, this results in an exhaustion of the normal hematopoietic system; in hematological malignancies, elevated concentration of IL-6 may lead to increased proliferation of leukemic cells 93 .
These data demonstrate that the hematopoietic niche in hematological malignancies is modified to the advantage of leukemic stem cells that are continuously fed and protected by the neighboring stromal cells; indeed, several preclinical studies are directed toward the mobilization of leukemic stem cells from their protective niche 94 .
The role of inflammation was also evaluated by studying an MDS murine model, in which a modification of MSCs was sufficient to establish a chronic pro-inflammatory BM microenvironment 22 . It has been shown that some inflammatory signals derive specifically from the mesenchymal compartment and may create a premalignant microenvironment increasing mutation rate in HSC and facilitating disease initiation. In more detail, the damage associated molecular pattern molecules, S100A8 and S100A9, secreted by altered MSCs drive mitochondrial dysfunction, oxidative stress, and DNA damage response in HSC, predisposing the BM to leukemic evolution. Coherently with these data, an activation of p53 tumor suppressor pathway, consequently to DNA damage in MSC-MDS, has been reported, from both in vitro and in vivo models 68 .
Beside cytokines, miRNA and extracellular vesicles (EVs) are topics of increasing interest among secreted factors for their capacity to influence the neighboring cellular population both at functional and molecular levels. EVs can be classified for size and origin. They can be separated in exosomes, small vesicles (40 to 100 nm) released as the consequence of multivesicular bodies fusion with plasma membrane, and microvesicles, bigger in size (50 to 1,000 nm) and secreted by cells as plasma membrane blebs 95 . In healthy conditions, a therapeutic potential of MSC-derived EVs has been widely demonstrated in both in vitro and in vivo models 96,97 . Data obtained from murine models showed that MSC-EVs can mitigate radiation injury in the BM, improving HSC proliferation and differentiation and accelerating their engraftment. This recovery may be exerted by proteins, nucleic acids, lipids, and metabolites contained in EV, but the basic biological mechanisms of their positive paracrine effect still need to be elucidated.
On the other side, this supporting capacity of MSC secretome may have a negative consequence in pathological condition; indeed, it was demonstrated that EV isolated from a serum deprived culture of MSC carry antiapoptotic, proliferative proteins, and tumor sustaining miRNA 98 . Accordingly, EV isolated from primary cultures of MSC-MDS has been demonstrated to harbor 21 miRNA upregulated in donor, and it was shown that these miRNA can be incorporated within HSC in vitro 72 . This mechanism produces an upregulation of miRNA 10a and miRNA 15a in the target cells that in turn lead to p53 gene overexpression 73 . This dysregulation in the miRNA content is related to the altered expression of endonucleases DICER and DROSHA. These genes encode for 2 endonuclease that regulate miRNA biogenesis, and it has been shown that a deletion of the RNase III Dicer1 in mesenchymal osteolineage cells is sufficient to produce a murine model with altered MSC osteogenic differentiation capability, altered texture of the bone matrix, and a significantly decreased number of osteoblasts 74 .
This animal model, even without any specific genetic alteration of HSCs, presented peripheral cytopenia with dysgranulopoiesis and marked dysplasia both in peripheral blood and in the BM consistently with MDS diagnosis. These data strongly suggested that an alteration in HSCs compartment might be sufficient to initiate the pathogenesis of MDS. Transplantation experiments performed in this work further confirmed the involvement of microenvironment in the disease insurgence; indeed, when leukemic HSCs from mutant mice was transplanted into wild-type animals, no signs of disease were assessed, but when wild-type HSCs were transplanted in lethally irradiated mutants, they developed leukopenia, anemia, and thrombocytopenia. These results show that hematopoietic progenitor cells themselves are not able to trigger the MDS pathogenesis, but they likely require a permissive microenvironment that supplies the needs for malignant HSC clones to propagate.
Consistent with this hypothesis, several attempts to propagate human MDS-HSC in animals to develop xenograft models just transplanting malignant hematopoietic progenitor clones did not yield satisfactory results 99 –101 .
Recently, another work of Medyouf and colleagues demonstrated, after having performed transplantation experiments, that the presence of MSC-MDSs improves the engraftment of HSC-MDSs in mice, confirming the necessity of a favorable microenvironment for the expansion of the disease 75 . However, it has also been shown that the injected MSCs remained in the recipient mice only 4 wks and localized in the injection site, contrary to HSC-MDS, which was able to expand and establish the disease also in noninjected bones. This observation (further confirmed by an in vitro coculture experiment performed by the same author) suggests that some HSC-MDSs after initial engraftment could be able to reprogram the BM niche and modify the mesenchymal stromal compartment to reconstruct a permissive microenvironment suitable for disease progression 102 . This hypothesis was further confirmed studying healthy MSCs after the exposure to HSC-MDS cells. It has been observed that after coculture with malignant HSCs, MSCs increased the expression of leukemia inhibitory factor; and the analysis of primary MSC-MDS transcriptome revealed that 1,008 genes were differentially expressed in donors and presented a significant enrichment in genes involved in response to an inflammatory environment.
This model really stresses the importance of mesenchymal compartment in MDS but raises the question of which is the first responsible of the normal cross-talk disruption among the cells of the niche. Studies of MSC isolated from patients after therapeutic treatment revealed that cells recover their physiological properties in patients who reached complete hematological remission.
In more detail, the hematopoietic supporting capacity, the proliferation capability, and gene expression of cells seem to be restored to the level of healthy MCSs 67,103 .
These data, assumed that the effect of the therapy is directed only toward HSC compartment, suggest that leukemic cells are able to modify the BM microenvironment taking advantage on normal HSC; however, it cannot be excluded that a pharmacological effect is exerted also on MSCs. It has been shown that the methylation levels of MSC-MDSs decrease after treatment with demethylating agents67; therefore, the improved outcome observed in patients could be the consequence of a therapeutical synergic effect on both stromal compartment and HSCs.
Balderman and colleagues showed how in a murine model of MDS the hematopoietic function is improved normalizing the microenvironment 23 . They observed that transgenic mice beside defect of HSCs presented increased numbers of nonfunctional MSCs and higher level of CCL3 and Vascular endothelial growth factor (VEGF) similarly to what was observed in human MDSs 104,105 . In addition, the exposure of HSC-MDS to a wild-type microenvironment improved the hematopoietic function in wild-type animals, demonstrating that a normal microenvironment can reduce the negative effect of pathological cells. On the other hand, if the MDS engraftment is high, the exposure to a healthy microenvironment is not sufficient to improve cytopenias, but it is more likely that leukemic cells affect the BM, which becomes supportive for leukemic HSC more than for healthy HSCs.
Such information becomes critical in consideration of future therapeutic approaches able to target both HSC and BM microenvironments. Several preclinical and early phase studies are directed toward the inhibition of CXCR4/CXCL12 axis between stroma and AML cells to mobilize leukemic cells out of their protective niche depriving the malignant clones from the MSC-derived survival signals 94 .
In conclusion, all data coming from in vitro and in vivo studies revealed that MDS pathogenesis results from a complex interaction between stromal and hematopoietic elements that together are able to instruct and modify their surrounding microenvironment. It is therefore assumable that, even if further studies are required, new strategies that develop synergic therapies directed both toward stromal and hematopoietic compartments are needed.
Footnotes
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.