
Abstract
Introduction
This study aimed to investigate the mechanisms underlying Tripterygium glycosides (TG) - induced liver injury, focusing on the role of the STING-TBK1 signaling pathway, and to evaluate the therapeutic potential of inhibiting this axis in mitigating liver damage.
Methods
The study employed two experimental approaches. First, male Balb/c mice were administered TG at doses of 13.5, 40.5, and 135 mg/kg for 3 weeks to assess dose-dependent hepatotoxicity. Liver injury was evaluated through serum ALT/AST levels, hepatic histopathology, and liver index. Immunohistochemical staining and Western blot analysis were used to examine STING expression in liver tissues and THP-1 cells. In the second approach, pharmacological inhibitors of STING and TBK1 were administered to evaluate their protective effects against TG-induced liver injury.
Results
TG induced dose-dependent liver injury and inflammatory infiltration, along with activation of the STING-TBK1 pathway in non-parenchymal cells. Inhibition of this pathway significantly attenuated hepatotoxicity, as evidenced by reduced ALT/AST levels, decreased inflammatory cytokines, and improved histopathological outcomes.
Discussion
These findings demonstrate that the STING-TBK1 axis plays a critical role in mediating TG-induced hepatotoxicity. Pharmacological inhibition of this pathway effectively alleviates TG-induced hepatotoxicity, suggesting its potential as a therapeutic target for drug-induced liver injury.
Introduction
Tripterygium glycosides (TG) are total glycosides extracted from Tripterygium wilfordii Hook. f. (TwHF). TG tablets are a traditional Chinese medicine formulation, which has been widely used in Chinese clinics for its anti-inflammatory and immunosuppressive effects. It has significant therapeutic effects on rheumatoid arthritis, systemic lupus erythematosus, nephrotic syndrome, anaphylactoid purpura, psoriasis and other diseases.1–3 However, the clinical application of TG is constrained by its narrow therapeutic window and dose-limiting toxicity.4,5 Liver injury is the most common toxicity of TG and has a high incidence. The main pathological manifestation of hepatotoxicity is acute liver injury. Patients typically present with gastrointestinal symptoms, and blood biochemical analysis reveals elevated levels of alanine aminotransferase (ALT) and aspartate aminotransferase (AST). 6 Consequently, a comprehensive understanding of the hepatotoxicity mechanism of TG is of great significance for the safe and rational use of TG in clinical practice.
The stimulator of interferon genes (STING) pathway has emerged as a crucial component of the innate immune system. Pathogen invasion triggers activation of the STING pathway, leading to phosphorylation of interferon regulatory factor 3 (IRF3) and subsequent production of interferon-β (IFN-β) and other cytokines that mediate immune defense.7–9 STING-mediated IFN-β secretion activates the JAK/STAT signaling cascade through interferon receptor binding, resulting in the production of inflammatory chemokine. 10 In addition, activation of STING promotes NF-κB signaling, further amplifying the inflammatory responses. 11 These properties establish the STING pathway as a promising therapeutic target for immune-related diseases.12,13 Although it is known that hepatic STING activation triggers inflammatory signaling and that TG-mediated hepatotoxicity is associated with oxidative stress, inflammation, and CYP450 enzyme regulation,14–16 the potential involvement of STING-mediated immune processes in TG-induced liver injury remains to be explored.
In this study, we investigated the functional role of the STING-TBK1 pathway in mediating TG-induced liver injury and evaluated the therapeutic potential of targeting this pathway using specific STING (H-151) and TBK1 (BX795) inhibitors. Results showed that TG activates the STING-TBK1 signaling pathway in hepatic nonparenchymal cells, which is a novel mechanism of TG-induced hepatotoxicity. Importantly, pharmacological inhibition of the STING-TBK1 axis by STING and TBK1 inhibitors attenuated TG-mediated liver injury and the associated inflammatory responses, suggesting the STING-TBK1 axis is a potential therapeutic target for preventing TG-related hepatotoxicity.
Materials and methods
Experimental animals
Two batches of male SPF mice aged 6-8 weeks were obtained from Chengdu Dashuo Laboratory Animal Co., LTD. Only male mice were used in this study to minimize hormonal effects on immune and liver responses. For the studies of TG-induced hepatotoxicity and STING/TBK1 inhibitor intervention experiment, mice were reared on a standard diet in the animal room of the department of pharmacology of Chinese Medicine, Shaanxi University of Chinese Medicine. Room temperature was maintained at a moderate level with adequate air exchange and appropriate humidity. Health status was monitored twice daily. Humane endpoints (including >20% weight loss or severe lethargy) warranted immediate euthanasia by CO2 asphyxiation.
All animal experimental procedures were conducted in accordance with ARRIVE guidelines and approved by the Animal Experiment Ethics Committee of the Shaanxi University of Chinese Medicine (Xianyang, China) (Ethics approval number: SUCMDL20230625006). Mice were monitored daily for signs of distress. Humane endpoints were established based on predefined clinical criteria. Animals were randomly assigned to experimental groups using a computer-generated random number sequence by an investigator not involved in outcome assessment. Group allocation was concealed until the completion of data collection. All outcome assessments, including histopathological scoring and biochemical measurements, were performed by investigators blinded to treatment allocation.
Experimental reagents
TG tablets were purchased from Zhejiang Deende Pharmaceutical Co., LTD. In our previous study, the chemical characterization of TG, including the quantitative components, has been fully characterized by UPLC-TQ-MS/MS. 17 ALT and AST kits were purchased from Nanjing Jiancheng Institute of Biological Engineering. RPMI1640 medium was purchased from Wuhan Boster Biological Engineering Co., LTD. Trypsin was purchased from Beijing Solarbio Biotechnology Co., LTD. Anti-STING, anti-TBK1 and anti-pTBK1 monoclonal antibodies were purchased from Cell Signaling Technology. Anti-β-actin was purchased from Abclone, and all secondary antibodies were purchased from Wuhan Boster Biological Engineering Co., LTD.
Animals treatment
To study of TG-induced hepatotoxicity, Balb/c male mice were randomly divided into four groups (n = 6 for per group) and acclimatized for 1 week: Control group (0 mg/kg TG), Low-dose group (13.5 mg/kg TG), Middle-dose group (40.5 mg/kg TG), High-dose group (135 mg/kg TG). The experimental dose of 13.5 mg/kg represents the clinically equivalent dose in mice, while 40.5 mg/kg and 135 mg/kg are equivalent to 3 and 10 times the clinical dose, respectively. dose selection. These doses selection was based on the following considerations: (1) Our preliminary experiments demonstrated that these doses induced significant hepatotoxicity suitable for mechanistic investigation; and (2) The 3-weeks duration aligns with standardized protocols for chronic hepatotoxicity studies to properly mimic clinical progression. Mice were given intragastric injections of saline (control) or the indicated concentrations of TG solution daily for 3 weeks. Body weights were recorded 24 h after the last dose. After isoflurane anesthesia, blood samples were collected for serum biochemical analysis, followed by euthanasia of the mice. All endpoints were evaluated, including serum ALT/AST levels, liver histopathology, and STING pathway activation markers.
To study the STING/TBK1 inhibition assay, Balb/c male mice were randomly divided into four groups (n = 6 per group): Control group (vehicle only), TG group (40 mg/kg), TG + H-151 (10 mg/kg STING inhibitor), and TG + BX795 (10 mg/kg TBK1 inhibitor). Mice were injected intraperitoneally with H-151 or BX795 1 hour prior to intragastric administration of 40 mg/kg TG. All compounds were administered at a volume of 0.1 mL/10 g body weight. Animals were analyzed for endpoints 12 h after administration.
Biochemical analysis
To quantify serum ALT and AST levels, a 20 μL of matrix solution was aliquoted into a 96-well plate, followed by the addition of 5 μL of the serum sample, and the mixture was incubated for 30 min at 37°C. Subsequently, 20 μL of 2,4-dinitrophenylhydrazine solution and 5 μL of sodium pyruvate standard solution (0.1 mmol/L) were added, and the reaction was continued for 20 min at 37°C. The reaction was terminated by adding 200 μL of 0.4 mol/L sodium hydroxide solution, followed by incubation for 15 min at room temperature. The absorbance was measured using a BioTek Synergy H1 Multimode microplate reader.
H&E staining
Fixed liver tissue was dehydrated, embedded and sectioned. Sections were stained with hematoxylin and eosin, then dehydrated and made transparent, followed by sealing with neutral gum. Inflammatory cells, steatosis and other lesions of the liver tissues were observed with microscope.
Immunohistochemistry (IHC)
Paraffin sections of liver tissues were dewaxed and washed with Phosphate buffered saline (PBS) after antigen repair, soaked in hydrogen peroxide to block endogenous peroxidase, and then sealed with serum. STING antibody was incubated overnight at 4°C, rinsed with PBS, and the secondary antibody was added. After incubation at 37°C for 30 min, the sections were stained with diaminobenzidine (DAB). When the positive part was brown-yellow, the sections were washed with distilled water, and the nuclei were re-stained with hematoxylin, with the sections effectively sealed with neutral gum. The expression level and expression site of STING protein were further observed by microscope.
Western blot
Protein samples were extracted from liver tissues (30 mg samples homogenized in RIPA buffer) or cultured THP-1 cells (1 × 106 cells per treatment). Following homogenization or lysis, the samples were centrifuged at 12,000×g for 15 min at 4°C. The supernatants were collected and protein concentrations were quantified using the BCA Protein Assay Kit. Samples were then added to the loading buffer, denatured by boiling, and separated by SDS-PAGE gels. The proteins on the SDS-PAGE gel were transferred to a PVDF membrane. After that, the PVDF membrane containing the target proteins was placed in 5% milk and incubated for 2 h at room temperature. The PVDF membrane containing target proteins were incubated with primary antibody and placed at 4°C overnight. After primary antibody incubation, the PVDF membrane was washed three times with TBST, then the secondary antibody was added and incubated for 1 h at room temperature. PVDF membrane was washed three times with TBST and then immunodetection was performed using chemiluminescence substrates. Western blot quantification was performed by normalizing target protein band intensities (STING, pTBK1 and TBK1) to β-Actin levels on the same membrane. After background subtraction using ImageJ, the relative expression was calculated as (target protein density)/(β-actin density) for each sample.
RT-qPCR
Primer sequences used for real-time PCR.

Statistical analysis
All statistical analyses were performed using GraphPad Prism v9.0.1 software. Data normality was assessed by the Shapiro-Wilk normality tests, and homogeneity of variance was verified by Brown-Forsythe tests. Central tendency and dispersion were expressed as Mean ± standard deviation (Mean ± SD). Comparisons between groups were analyzed by one-way ANOVA followed by Levene’s test to verify homogeneity of variance. Statistical significance was defined as follows: *p < 0.05 and **p < 0.01 versus control group; #p < 0.05, ##p < 0.01 and ###p < 0.001 versus model group.
Results
TG induces dose-dependent liver injury and hepatic inflammation
As shown in Figure 1(a), 3-week TG treatment at different doses (13.5, 40.5, and 135 mg/kg) showed no significant change in the liver index in mice. However, serum AST levels were markedly elevated in the high-dose groups compared with the control group (Figure 1(b)). Similarly, serum ALT levels were significantly elevated in the high-dose group (Figure 1(c)), suggesting that TG induced liver injury. Histopathological analysis (Figure 1(d)) showed that control mice exhibited well-preserved hepatic architecture, whereas the low-dose group exhibited a slight inflammatory cell infiltration. In contrast, the middle- and high-dose groups showed pronounced inflammatory infiltration along with an increase in hepatocyte vacuoles, indicating progressive hepatocyte injury and steatosis. The results demonstrate that TG triggers dose-dependent liver injury as evidenced by elevated transaminase levels, inflammatory infiltration, and hepatocellular degeneration. TG-induced hepatotoxicity and inflammation. Control group refers to the normal group given 0.9% physiologic saline. “low” refers to the administration of TG at 13.5 mg/kg; “middle” refers to the administration of TG at 40.5 mg/kg; “high” refers to the administration of 135 mg/kg of TG. (a) Liver index of mice after 3 weeks of TG treatment (n = 6 per group). Liver index = (liver weight/body weight) × 100%; (b) Serum AST levels post-TG administration (n = 6); (c) Serum ALT levels post-TG administration (n = 6); (d) Representative images of hematoxylin and eosin staining. Red arrow represents to inflammatory infiltration; blue arrow indicates to hepatocyte vacuolation and steatosis. Scale bar = 100 μm. *p < 0.05 and ****p < 0.0001 versus control. AST: Aspartate aminotransferase; ALT: Alanine aminotransferase.
TG activates the STING-TBK1 axis in hepatic non-parenchymal cells
To elucidate the role of STING-TBK1 pathway in TG-induced hepatotoxicity, we analyzed liver tissues using immunohistochemistry (IHC) and Western blot. The results showed that STING expression was significantly elevated in TG-treated livers (Figure 2(a)–(d)) and was mainly localized in non-parenchymal cells rather than hepatocytes (as indicated by red arrows in Figure 2(a)). Then, the activation of STING-related signaling pathways was further investigated by applying THP-1 cells, a mononuclear cell line isolated from the peripheral blood of patients with acute monocytic leukemia. They are suspension cells that have been widely used to study monocyte/macrophage functional mechanisms, signaling pathways, drug transport and immune responses. In addition, THP-1 cells are endogenous immune cells that express STING, facilitating the study of drug effects on the STING pathway. Triptolide (TPL) was identified as the major active ingredient of TG.
18
Consistent with our in vivo findings, TPL treatment markedly increased the phosphorylation of TBK1 (Figure 2(e)–(f)), confirming the activation of downstream pathway. Together, these data demonstrate that TG-induced liver injury involves selective activation of the STING-TBK1 axis in hepatic nonparenchymal cells, highlighting the critical role of innate immune signaling to TG-mediated liver injury. TG induced the activation of STING-TBK1 axis in liver non-parenchymal cells. (a) IHC staining of liver STING. Red arrow corresponds to brown DAB staining, indicating positive STING expression; (b) Quantification of the positive area of STING-stained tissue (n = 3); (c) Western blot of liver STING after TG treatment (n = 3); (d) Quantitative analysis of (c); (e) Western blot of STING, TBK1 and pTBK1 in THP-1 cells after TPL treatment; (f) Quantitative analysis of (e), Western blot statistics were applied to 3 technical replicates. *p < 0.05, **p < 0.01, ***p < 0.001 and ****p < 0.0001 versus control. DAB: Diaminobenzidine; STING: Stimulator of interferon genes; TBK1: TANK-Binding Kinase 1; pTBK1: Phosphorylated TANK-Binding Kinase 1; TPL: Triptolide.
Pharmacological inhibition of STING-TBK1 signaling attenuates TG-induced acute liver injury
Based on our findings that TG triggers STING-TBK1 activation predominantly in hepatic non-parenchymal cells (Figure 2), we hypothesized that pharmacological inhibition of this pathway would attenuate liver injury. To mechanistically validate this and assess the therapeutic potential, we selected two specific inhibitors (Figure 3(a)). H-151 is a covalent STING inhibitor that blocks palmitoylation of STING. BX795 is a TBK1 inhibitor known to suppress STING downstream signaling. Before and after 12 h of intervention, the body weight of all groups of mice, including the control group, decreased slightly (Figure 3(b)). As shown in Figure 3(c), the liver index of mice in the TG intervention group was significantly increased, while those of mice in the H-151 co-intervention group were significantly lower. Biochemical assays showed that the levels of ALT and AST were significantly elevated in the TG group of mice, whereas those of the H-151 group of mice were significantly reduced (Figure 3(d) and (e)). In addition, BX795 also significantly reduced AST levels but had no significant effect on liver index and ALT levels, suggesting that H-151 was more effective than BX795. These findings provide compelling evidence that TG-mediated acute liver injury can be effectively ameliorated by pharmacologic disruption of the STING-TBK1 axis, particularly STING levels. STING-TBK1 inhibitor alleviates TG-induced hepatotoxicity. Control group refers to the normal group given 0.9% saline. Model group refers to the administration of TG; H-151 refers to the administration of TG and H-151; BX795 refers to the administration of TG and BX795; (a) Flow chart overview of STING/TBK1 inhibition study; (b) Body weight changes in mice (n = 6 for biological replicates); (c) Liver index of mice after TG and H-151 or BX795 treatment (n = 6 for biological replicates); (d) Serum ALT levels in different groups (n = 6) for biological replicates; (e) Serum AST levels in different groups (n = 6 for biological replicates). **p < 0.01, ***p < 0.001 and ****p < 0.0001 versus control. #p < 0.05, ##p < 0.01 versus model group. TG: Tripterygium glycosides; ALT: Alanine aminotransferase; AST: Aspartate aminotransferase.
Pharmacological inhibition of STING-TBK1 signaling attenuates TG-induced inflammatory responses
To investigate the anti-inflammatory effects of STING-TBK1 pathway inhibition, we analyzed hepatic inflammatory factors. As shown in Figure 4(a)–(c), ELISA analysis revealed that both H-151 (STING inhibitor) and BX795 (TBK1 inhibitor) significantly reduced serum levels of pro-inflammatory cytokines (TNF-α, IL-1β, and IL-6) when compared to TG-treated controls (Figure 4(a)–(c)). In addition, RT-qPCR analysis demonstrated significant downregulation of IFN-β, IL-1β, and IL-6 mRNA levels in liver tissues of the inhibitor-treated groups (Supplemental Figure S1). The consistent suppression of both cytokine secretion and gene expression confirms the crucial role of STING-TBK1 signaling in TG-induced inflammation. These results suggest that targeted inhibition of the STING-TBK1 signaling pathway attenuates TG-induced liver inflammation. Therefore, TG may increase the expression of IFN-β and inflammatory factors by activating STING-TBK1 signal in liver non-parenchymal cells, thus leading to inflammatory response and liver injury. By targeting inhibition of STING and TBK1, liver inflammation and injury can be attenuated (Figure 5). STING-TBK1 inhibitors reduced TG-induced liver inflammation; (a) Serum TNF-α levels measured by ELISA assay (n = 6); (b) Serum IL-1β levels measured by ELISA assay (n = 6); (c) Serum IL-6 levels measured by ELISA assay (n = 6). *p < 0.05 and **p < 0.01 versus control. #p < 0.05, ##p < 0.01 and ###p < 0.001 versus model group. TNF-α: Tumor Necrosis Factor-α; IL-1β: Interleukin-1β; IL-6: Interleukin-6. STING-TBK1 inhibitors alleviated TG-induced IFN-β activation and inflammatory response. TG may induce the expression of IFN-β and inflammatory factors by activating STING-TBK1 signal in liver non-parenchymal cells, thus leading to inflammatory response and liver injury. H-151 and BX795 could reduce liver inflammation and injury by targeting inhibition of STING and TBK1. STING: Stimulator of interferon genes; TBK1: TANK-Binding Kinase 1; pTBK1: Phosphorylated TANK-Binding Kinase 1; IKK: Inhibitor of kappa B kinase; NF-κB: Nuclear factor kappa-B; IRF3: Interferon regulatory factor 3; IFN-β: Interferon-beta; TNF-α: Tumor Necrosis Factor-α; IL-1β: Interleukin-1β; IL-6: Interleukin-6.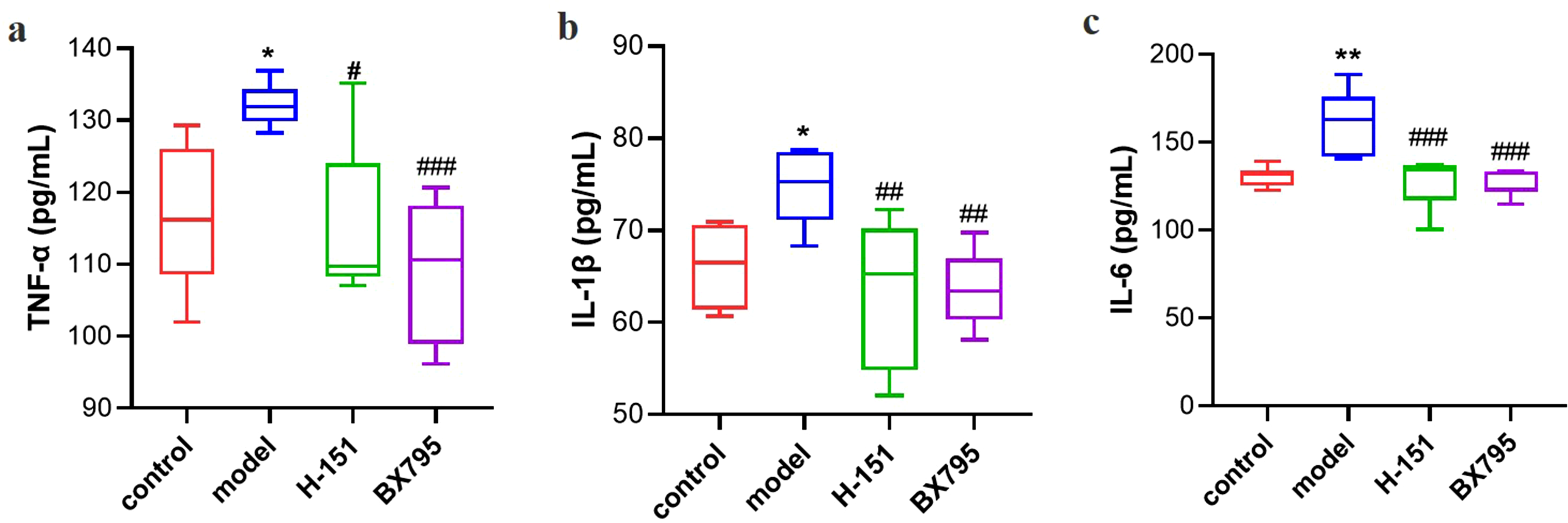

Discussion
Despite its efficacy in rheumatoid arthritis and other immunoinflammatory diseases, the clinical application of TG is limited by hepatotoxicity, the mechanism of which is not fully understood. In this study, we found that serum AST and ALT levels were significantly elevated in mice treated with TG, suggesting that TG can induce significant liver injury. Histopathological examination by H&E staining further demonstrated that TG-induced liver injury was accompanied by inflammatory infiltration and cell steatosis in a dose-dependent manner. Although activation of the STING pathway has been associated with organ toxicity, including triptolide-induced nephrotoxicity and bleomycin-induced pulmonary fibrosis,19,20 its involvement in TG-mediated hepatotoxicity had not been previously investigated. To elucidate the underlying mechanisms, we performed immunohistochemical and western blot analyses, which showed that STING protein expression was markedly upregulated, especially in hepatic nonparenchymal cells. Given that macrophages represent a key non-parenchymal cell population, we employed THP-1 cells (a human monocyte model) to assess activation of the STING pathway. Notably, TG treatment induced TBK1 phosphorylation in THP-1 cells, thus providing compelling evidence that TG-induced hepatotoxicity involves activation of the STING-TBK1 signaling axis in liver non-parenchymal cells.
Given the pivotal role of STING-TBK1 signaling in TG-induced hepatotoxicity, we investigated the therapeutic potential of targeted pathway inhibition. We employed H-151, a covalent STING inhibitor known to suppress STING-mediated inflammatory cytokine production in human and murine cells, which has shown significant efficacy in ameliorating autoinflammatory pathology in mouse models. 21 Recent studies have shown that H-151 can attenuate kidney damage caused by sepsis via inhibition of STING-TBK1 signaling activation. 22 BX795 is a potent TBK1 inhibitor that inhibits the phosphorylation activation of TBK1 kinase, thereby blocking the phosphorylation, nuclear translocation and transcriptional activity of IRF3.23,24 To our knowledge, this is the first in vivo evidence that STING-TBK1 inhibitors mitigate TG-induced liver injury. Our study suggests that targeted inhibition of the STING-TBK1 signaling pathway is a therapeutic strategy to attenuate the hepatotoxicity of TG.
However, the detailed molecular mechanisms and clinical translation potential remain incompletely understood. Mechanistically, we propose that TG may activate STING signaling through direct and indirect pathways: firstly, the main bioactive components of TG may directly bind to STING protein and then induce activation of STING-TBK1 signaling. Secondly, TG induces mitochondrial damage and oxidative stress, which disrupts mitochondrial membrane potential and leads to the release of mitochondrial DNA, thereby activating STING. In terms of clinical translation, while these findings identify novel therapeutic targets for clinical intervention, several critical challenges must be addressed. Foremost among these is the consideration of species-specific differences, as human STING exhibits distinct ligand sensitivity patterns compared to murine STING. 25 In addition, While we highlight the shared STING-TBK1 pathway activation and inflammatory responses between THP-1 cells and hepatic macrophages, we now clearly acknowledge several critical limitations: (1) inherent differences between a monocytic leukemia cell line and primary hepatic macrophages in terms of metabolic profiles and polarization states; (2) potential variations in drug uptake and clearance mechanisms; and (3) the essential need for validation in primary murine liver non-parenchymal cells. We emphasize that while THP-1 studies provide valuable mechanistic insights, their findings must be interpreted in conjunction with physiological models.
Based on the limitations of the current study, we propose several major directions for future research. First, although the present study focused on male animals to control for sex variables, we acknowledge this as a limitation that needs further investigation. Our future research will focus on establishing female animal models to systematically examine potential sex differences in STING-TBK1 pathway activation and hepatotoxic responses and evaluating the efficacy of targeted therapeutic strategies across different sex models. Second, oxidative stress and mitochondrial DNA levels must be investigated to determine the mechanisms of TG-induced STING activation. Furthermore, to advance these findings toward clinical translation, future studies should focus on validating these mechanisms in patient-derived liver organoid models.
In conclusion, our study demonstrates that TG-induced hepatotoxicity is associated with the activation of the STING-TBK1 signaling pathway in liver non-parenchymal cells, especially macrophages. The therapeutic efficacy of STING and TBK1 inhibitors in our model suggests that targeting this pathway could be a viable strategy to mitigate TG-related hepatotoxicity. These approaches have the potential to expand the therapeutic window for TG in autoimmune diseases where current dosing is limited by hepatotoxicity concerns. However, the sensitivity of human STING to ligands is different compared to murine STING, necessitating validation in humanized models. Future studies should focus on validating these findings in primate models or clinical trials. These findings provide a mechanistic basis for improving the safety of TG while highlighting critical gaps for clinical translation.
Supplemental Material
Supplemental Material - Targeted inhibition of STING-TBK1 axis alleviates the hepatotoxicity of tripterygium glycosides
Supplemental Material for Targeted inhibition of STING-TBK1 axis alleviates the hepatotoxicity of tripterygium glycosides by Ting Yang, Tian Ma, Huanhuan Zhang, Zechen Niu, Chunzhou Cai, Dongxiao Cui in Human & Experimental Toxicology
Footnotes
Ethical Considerations
All animal procedures were performed in accordance with the ARRIVE guidelines and approved by the Animal Experiment Ethics Committee of the Shaanxi University of Chinese Medicine (Xianyang, China) (Ethics approval number: SUCMDL20230625006). Mice were monitored daily for signs of distress. Humane endpoints were established based on predefined clinical criteria. Animals were randomly assigned to experimental groups using a computer-generated random number sequence by an investigator not involved in outcome assessment. Group allocation was concealed until the completion of data collection. All outcome assessments, including histopathological scoring and biochemical measurements, were performed by investigators blinded to treatment allocation.
Author Contributions
Ting Yang: Conceptualization, Formal analysis, Writing - original draft. Tian Ma and Huanhuan Zhang: Conceptualization, Formal analysis, Data curation. Zechen Niu and Chunzhou Cai: Investigation, Data curation, Visualization. Dongxiao Cui: Supervision, Project administration, Writing - review & editing.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: Natural Science Basic Research Program of Shaanxi (Program No. 2024JC-YBQN-0925). Xianyang Chinese Medicine Industrial Innovation Cluster Project of Qin Innovation Source (Program No. L2024-QCY-ZYYJJQ-X220).
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Data Availability Statement
Data will be made available on request.
Supplemental Material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.