
Abstract
Introduction
Cigarette smoking extract (CSE) can cause endothelial cell (EC) dysfunction, and then promote the occurrence and development of atherosclerosis. However, the molecular mechanisms underlying CSE-induced EC dysfunction are unknown. Sirt1, as a deacetylase, is involved in various biological processes of ECs. Therefore, this study investigated whether CSE induces apoptosis and mitochondrial dysfunction in human umbilical vein endothelial cells (HUVECs) via Sirt1-dependent mechanisms.
Methods
HUVEC activity was assessed using MTT and crystal violet staining following treatment with different concentrations of CSE. Lentiviral transfection technology was used to generate HUVECs overexpressing Sirt1. Apoptosis was detected by Tunnel staining. MitoTracker™ Deep Red FM and JC-1 were used to assess mitochondrial structure and membrane potential. ELISA was used to detect the expression of superoxide dismutase (SOD) and malondialdehyde (MDA). qPCR was used to determine mRNA expression. Atherosclerosis was evaluated by oil red O staining in ApoE-KO mice after cigarette smoke exposure.
Results
CSE decreased Sirt1 and sonic hedgehog (SHH) expression, leading to mitochondrial dysfunction and apoptosis in HUVECs. Overexpressing Sirt1 or activating the SHH signaling pathway attenuated CSE-induced apoptosis and mitochondrial dysfunction. However, inhibiting the SHH signaling axis attenuated the protective effect of Sirt1 overexpression on CSE-induced apoptosis and mitochondrial dysfunction. In vivo studies also showed that cigarette smoke exacerbated atherosclerosis in ApoE-KO mice, downregulating Sirt1, SHH, and Gli1 expression in the aorta. Additionally, cigarette smoke increased Bax expression and decreased Bcl-2 expression in ApoE-KO mice aortas.
Discussions
Smoking can affect all stages of the atherosclerosis process, and the specific mechanism remains unclear. This study confirms that CSE can induce mitochondrial dysfunction and apoptosis of HUVECs by reducing Sirt1 expression and inhibiting SHH signaling activation. These findings provide new insights into the prevention and treatment of smoking-induced atherosclerosis.
Introduction
The progressive population aging and lifestyle changes have contributed to the increased prevalence of cardiovascular diseases, endangering human health.1,2 Atherosclerosis is the common underlying pathology, but its etiology and pathogenesis are complex and multifactorial. 3
Endothelial cells (ECs) are the main cells in the intimal layer of arteries, and alterations in their function compromise the arterial intima barrier. Cigarette smoking extract (CSE) can cause EC dysfunction, serving as a prerequisite and initiating factor for atherosclerosis.4,5 CSE contains numerous harmful substances, such as nicotine, oxygen-free radicals, and carbon monoxide. 6 It can damage ECs through various mechanisms, thereby causing EC dysfunction.7,8 However, the molecular mechanisms underlying CSE-induced EC dysfunction are still unclear.
Sirt1 is a deacetylase dependent on nicotinamide adenosine dinucleotide (NAD+). It promotes the deacetylation of substrate proteins and regulates DNA expression, cell apoptosis, aging, and other processes.9–11 However, it remains unclear whether CSE induces mitochondrial dysfunction and apoptosis in ECs through Sirt1-dependent mechanisms.
Materials and methods
Antibodies and reagents
Antibodies Sirt1 and GAPDH were purchased from Santa Cruz. Bax, Bcl-2, Gli1, SMO, and SHH were purchased from Abcam. SAG and HPI-4 (Ciliobrevin A) were obtained from Selleck (USA). JC-1, MDA, and SOD were purchased from Beyotime. The primers were prepared by the Beijing Genomics Institution (BGI). Crystal Violet was obtained from Amresco/Sigma.
CSE preparation
CSE was prepared by removing the filter from the cigarette (Marlboro), lighting it, and collecting the smoke in the DMEM medium using a smoke recovery device. The combustion control time for each cigarette was approximately 3 min. The resulting smoke stock was filtered through a 0.22 µm filter, yielding a 100% concentration of smoke stock.
Cell culture
Human umbilical vein endothelial cells (HUVECs) were obtained from the American Type Culture Collection (ATCC, Manassas, VA). DMEM medium (Gibco) containing 10% fetal bovine serum (FBS) and 1% penicillin-streptomycin was used. The cells were cultured in a 95% air /5%CO2 cell incubator at 37°C.
Cell viability assays
Cell activity was detected using the MTT(3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2-H-tetrazolium bromide) assay. In brief, 2000 cells were inoculated in 96-well plates and treated under different conditions. After the intervention, 10 µL MTT was added to each well, followed by incubation at 37°C for 4h. The medium was then discarded, and 100 µL DMSO was added to each well. After shaking for 15 min, the absorbance was measured using a microplate analyzer at a wavelength of 570 nm.
Quantitative real-time PCR (qPCR)
Cells were collected and RNA was extracted. The RNA was then reverse-transcribed into cDNA for qPCR analysis using the Revertra Ace® kit (Toyobo, Japan), according to the manufacturer’s instructions. The qPCR reaction mixture was prepared using the SYBR Green qPCR Master Mix kit, and amplification was performed on an Applied Biosystems ABI Viia7 instrument (Thermo Fisher Scientific, USA). The following primer sequences were used: Sirt1 (5-TAGCCTTGTCAGATAAGGAAGGAGAGA-3 and 5-ACAGCTTCACAGTCAACTTTGT-3), GAPDH (5- CAGTCTTCTGGGTGGCAGTGA -3 and 5- TGCACCACCAACTGCTTAGC -3)
Lentivirus transfection
Cells were seeded in 6-well plates at a concentration of 2 × 105 cells per well. Lentivirus suspension, diluted in culture medium to a multiplicity of infection (MOI) of 10, was added to each well. After incubation for 24 h, the culture was continued in a fresh medium. Stably transfected lentivirus cells were established after puromycin screening.
ELISA detection
HUVECs were seeded in 6-well plates. After the intervention treatment, the cells were collected, lysed, and centrifuged to obtain the supernatant. The malondialdehyde (MDA) and superoxide dismutase (SOD) contents were determined according to the kit instructions (Beyotime).
TUNEL assay
2 × 105 cells were inoculated in 6-well plates and treated with different conditions. Cell Apoptosis was detected according to the instructions of the kit (One Step TUNEL Apoptosis Kit, Beyotime) and photographs were obtained using an inverted fluorescent microscope.
Mitochondrial assay
For mitochondrial staining, the cells are first seeded in a petri dish (BeyoGold™ 35 mm Confocal Dishes, Beyotime). After the intervention, the medium was aspirated and MitoTracker™ Deep Red FM (Thermo Scientific) was added for staining. Mitochondrial membrane potential was detected after the intervention according to the kit instructions (JC-1, Beyotime). Stained cells were visualized using a confocal laser microscope (Leica TCS SP8, Germany).
Immunofluorescence
Cellular immunofluorescence: Cells were seeded on coverslips in a 6-well plate. After intervention, cells were fixed, permeabilized, and incubated overnight with the primary antibody. Cells were then incubated with fluorescently labeled secondary antibodies at 37°C for 1 h in the dark, followed by DAPI staining for 15 min. Coverslips were mounted with an antifade mounting medium (Beyotime) and photographed using a confocal laser microscope (Leica TCS SP8, Germany).
Tissue immunofluorescence: Extracted vascular tissue was fixed with 4% paraformaldehyde, embedded, and processed into paraffin blocks. Tissue sections were deparaffinized, blocked, and incubated overnight with primary antibodies. Secondary antibodies were then applied, followed by DAPI staining. Sections were mounted with an antifade mounting medium (Beyotime) and photographed using a confocal laser microscope (Leica TCS SP8, Germany).
Western blot assay
After the intervention, cells were collected, lysed, and centrifuged, and the supernatant was extracted. Proteins were denatured and separated by SDS-polyacrylamide gel electrophoresis using a concentration appropriate for the molecular weight of the target protein. Proteins were then transferred to a PVDF membrane. After incubating with 0.5% BSA for 1 h, the PVDF membrane was placed in the primary antibody and incubated overnight at 4°C. After combining the fluorescently labeled secondary antibody with the primary antibody, Li-COR Odyssey infrared fluorescence scanning was used for imaging.
Animal experiments
Twenty 8-week-old (20 ± 5 g) male ApoE knockout (ApoE-KO) mice were purchased from Changzhou Cavens Laboratory Animal Co. Ltd. The mice were housed in the SPF room of the Nuclear Medicine and Molecular Imaging Key Laboratory of Sichuan Province, Affiliated Hospital of Southwest Medical University (Housing conditions: 12 h light/12 h dark cycle, temperature 20∼26°C, humidity 55%∼65%, ad libitum access to food and water). All mice experiments were in accordance with the Guide for the Care and Use of Laboratory Animals published by the National Institutes of Health (NIH Publication No. 85-23, revised in 1996) and approved by Southwest Medical University for laboratory animal ethics.
Mouse smoke exposure
Cigarette smoke (CS) exposure was performed as reported by Gairola CG et al. 12 Briefly, the exposure group consisted of ApoE-KO mice exposed to sidestream cigarette smoke (SSCS) for 6 h/day, 5 days/week, over 12 weeks. The control group underwent a similar procedure but was exposed to filtered fresh air. After 12 weeks of feeding, the mice were euthanized by cervical dislocation under pentobarbital sodium anesthesia, and the aortas were harvested.
Administration of animals
After a 1-week environmental adaptation period, 20 mice were randomly divided into two groups: (1) Control group (Con, n = 10); and (2) Cigarette smoke group (CS, n = 10). All mice received a high-fat diet (High Fat Clinton-Cybulsky Rodent Diet with 1.25% Cholesterol and 0.5% Sodium Cholate, Research Diets, USA). Body weight was measured weekly throughout the study.
Oil red O staining
After the sections were stained with oil red O, nuclei were counterstained with Sumu finishes. Following staining, slides were sealed with a water-sealing liquid. Images were then captured using a regular microscope equipped with a digital camera.
Electron microscopy
Tissues were prefixed with 3% glutaraldehyde, followed by postfixation in 1% osmium tetroxide. Samples were then dehydrated in a graded acetone series, infiltrated with Epox 812, and embedded. Semithin sections were stained with methylene blue and ultrathin sections were cut using a diamond knife and stained with uranyl acetate and lead citrate. Sections were examined using a JEM-1400-Flash transmission electron microscope.
Statistical analysis
Data were presented as mean ± standard deviation (SD). Differences between two groups were assessed using Student's t-test, while the differences between three or more groups were assessed using one-way analysis of variance (ANOVA). Add the post hoc test after ANOVA. SPSS 21.0 was used for statistical analysis; P < 0.05 was considered statistically significant.
Results
CSE induces HUVEC apoptosis and mitochondrial dysfunction
CSE is a liquid mixture collected from cigarettes after burning and filtering. It contains thousands of harmful substances that can affect cell growth through various mechanisms. To determine the optimal concentration and time point for CSE intervention, HUVECs were treated with CSE at different concentrations (0%, 1%, 2%, 4%, 8%, 10%, 15%, 20% and 30%) for 24 h, 48 h and 72 h. Cell activity was assessed using the MTT assay (Figure 1(a)). Crystal violet staining was used to assess the growth of HUVECs after exposure to various concentrations of CSE for 24 h (Figure 1(b)–(c)). Results showed that 4% CSE did not significantly impact cell activity after 24 h of exposure. Based on these findings, 4% CSE for 24 h was used as the optimal condition for subsequent experiments. CSE induces HUVEC apoptosis and mitochondrial dysfunction. After treatment with different concentrations of CSE, the activity of HUVECs (a) was measured by MTT assay, and crystal violet staining was used to assess the growth of HUVECs (b, c). (d) Detection of HUVEC apoptosis by Tunnel staining. Western blot (e, f) and immunofluorescence (g, h, i; magnification: ×1200) assays to detect the expression of apoptosis-related proteins Bax and Bcl-2. (j) MitoTracker™ Deep Red FM staining of mitochondria. JC-1 for detecting mitochondrial membrane potential (k, l; magnification: ×800). Results of ELISA showing the expression levels of MDA (M) and SOD (N). *p < 05, **p < 01, ***p < 001.
To further elucidate the effects of CSE on HUVEC apoptosis and mitochondrial function, we treated HUVECs with 4% CSE for 24 h and then tested the relevant indicators. The results showed that compared with the control group, apoptosis was significantly increased in the 4% CSE intervention group (Figure 1(d)). Western blot (Figure 1(e)–(f)) and immunofluorescence (Figure 1(g)–(i)) assays also showed downregulation of the apoptosis-related protein Bcl- 2 and upregulation of Bax. Further, compared with the control group, the mitochondrial structural integrity of the 4% CSE intervention group was poor, and the fluorescence density was significantly weakened (Figure 1(j)). HUVECs treated with 4% CSE showed increased green fluorescence expression and decreased mitochondrial membrane potential (Figure 1(k) and (l)). ELISA assay showed that 4% CSE increased MDA expression (Figure 1(m)) and decreased SOD expression in HUVECs (Figure 1(n)). These findings indicate that CSE can promote apoptosis and mitochondrial dysfunction in HUVECs.
CSE can reduce Sirt1 expression and inhibit the SHH signal pathway
Sirt1 is a class of NAD+-dependent histone deacetylases widely found in living organisms. Numerous studies have shown that dysregulation of Sirt1 can affect the occurrence and development of a variety of diseases.
9
In our in vitro study, immunofluorescence assay showed that 4% CSE intervention in HUVECs for 24h significantly reduced Sirt1 expression compared with the control group (Figure 2(a) and (b)). In HUVECs, the expression of Sirt1 showed a progressive decrease after 4% CSE intervention at different time points (0h, 6h, 12h, 24h) (Figure 2(c)). Moreover, the Sirt1 expression in HUVECs decreased with the increase in CSE concentration at 24 h after treatment with different concentrations of CSE (Figure 2(d)). CSE reduces Sirt1 expression and inhibits the SHH signaling pathway. After treatment of HUVECs with 4% CSE for 24 h, immunofluorescence (a, b; magnification: ×1200) was used to detect Sirt1 expression. (c) After 4% CSE exposure of HUVECs, Sirt1 expression was detected by Western blot at different time points (0h, 6h, 12h, 24h). (d) After 24 h of exposure of HUVECs with different concentrations of CSE (0%, 1%, 2%, 4%), Sirt1 expression was detected by Western blot. Immunofluorescence (e–h) and Western blot (i–l) assays to detect the expression of SHH signaling pathway-related proteins SHH, SMO, and Gli1. *p < .05, **p < .01, ***p < .001.
The Sonic Hedgehog (SHH) signaling pathway regulates the differentiation and growth of several cell types and plays a key role in maintaining normal body functions. Several basic and clinical studies have shown that aberrant regulation of SHH signaling pathways causes atherosclerosis.13,14 We further investigated whether CSE can cause activation or inhibition of the SHH signaling pathway. Western blot assay showed that SHH signaling pathway-related proteins (Gli1, SMO, SHH) in HUVECs were significantly down-regulated at 24 h after 4% CSE intervention compared with the control group (Figure 2(i)–(l)). Immunofluorescence showed that the expression of SHH and SMO in the intervention group was consistent with the results of the Western blot assay (Figure 2(e)–(h)). These findings indicate that CSE can reduce the expression of Sirt1 and inhibit the SHH signal pathway.
High expression of Sirt1 can attenuate CSE-induced HUVEC apoptosis and mitochondrial dysfunction
We next investigated the potential involvement of Sirt1 in CSE-induced mitochondrial dysfunction and apoptosis in HUVECs. We constructed a lentivirus overexpressing Sirt1 (OE-Sirt1) and established stable Sirt1-overexpressing HUVECs (OE-Sirt1-HUVECs) after transfection (Figure 3(a) and (b)). Successful establishment of the OE-Sirt1-HUVECs model was verified by Western Blot (Figure 3(c) and (d)) and qPCR (Figure 3(e)) assays, confirming upregulated Sirt1 expression. After 4% CSE exposure for 24h, OE-Sirt1-HUVECs showed improved mitochondrial structural integrity and increased fluorescence density compared to HUVECs (Figure 3(f)). Additionally, OE-Sirt1-HUVECs exhibited decreased MDA expression (Figure 3(g)) and increased SOD expression (Figure 3(h)). Compared to HUVECs, the OE-Sirt1-HUVECs group showed downregulated Bax expression (Figure 3(i) and (k)), and upregulated Bcl-2 expression (Figure 3(j) and (l)). These findings suggest that Sirt1 overexpression reduces CSE-induced apoptosis and mitochondrial dysfunction in HUVECs. Sirt1 overexpression can attenuate CSE-induced mitochondrial dysfunction and apoptosis in HUVECs. (a) Structural illustration of the lentivirus overexpressing Sirt1; (b) After screening, the fluorescence of the stably transfected empty vector lentivirus and Sirt1 overexpression. Western blot (c, d) and qPCR (e) assays to detect Sirt1 overexpression. (f) MitoTracker™ Deep Red FM staining of mitochondria. The expression levels of MDA (g) and SOD (h) were detected by ELISA. Western blot assay to detect the expression of apoptosis-related proteins Bax (i, k) and Bcl-2 (j, l). *p < .05, **p < .01, ***p < .001.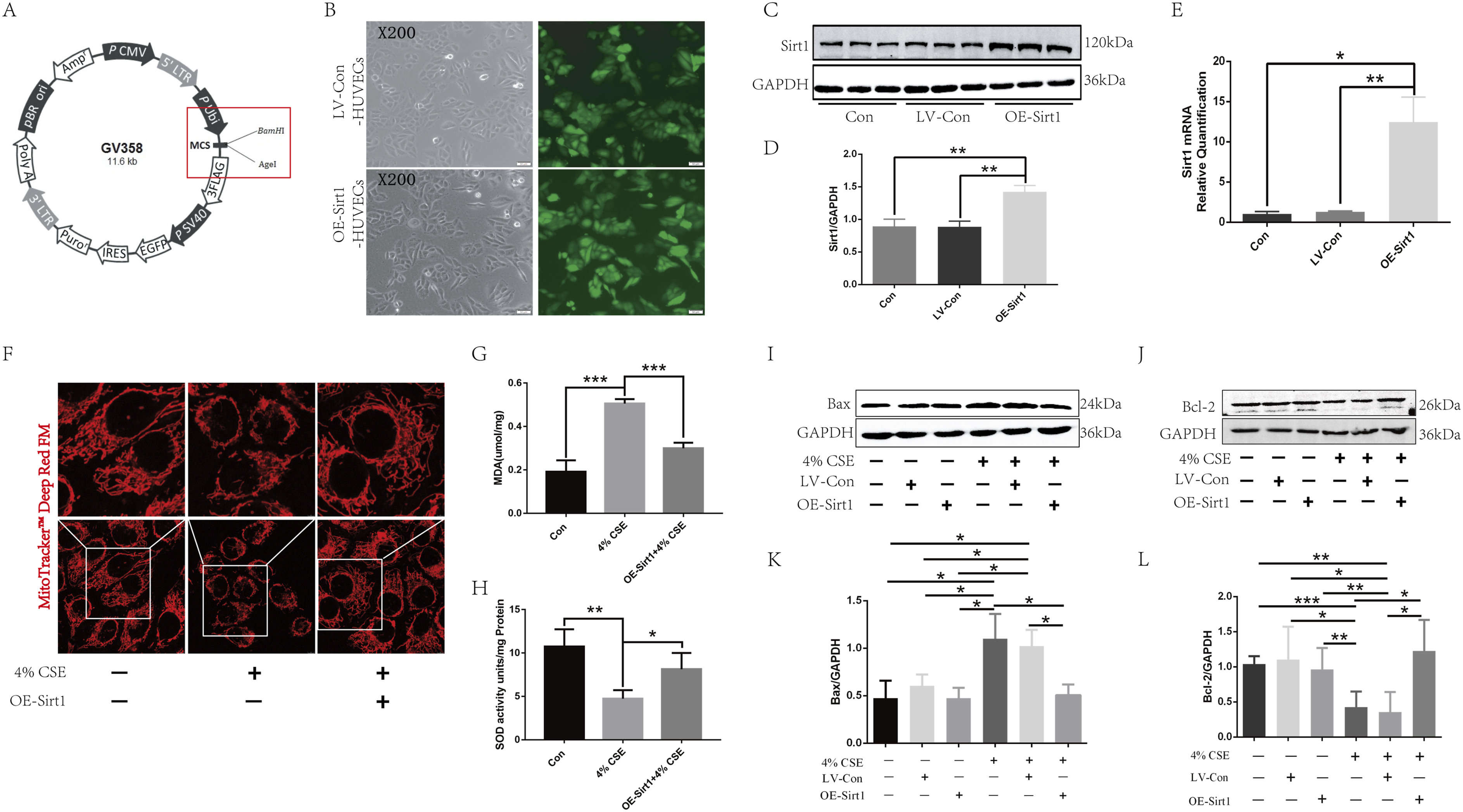
Sirt1-SHH axis is involved in CSE-induced apoptosis and mitochondrial dysfunction in HUVECs
We next investigated the potential involvement of the Sirt1-SHH axis in CSE-induced apoptosis and mitochondrial dysfunction in HUVECs. Treatment with SAG, an SHH signaling pathway agonist, significantly improved mitochondrial structural integrity (Figure 4(a)) and membrane potential (Figure 4(b) and (c)) in the SAG-treated HUVECs group compared to the untreated group. Additionally, SAG treatment decreased MDA expression (Figure 4(h)) and increased SOD expression (Figure 4(g)). Compared to the control group, SAG treatment downregulated Bax expression and upregulated Bcl-2 expression (Figure 4(d)–(f)). However, inhibition of the SHH signaling pathway using HPI-4 reduced mitochondrial structural integrity and fluorescence intensity in OE-Sirt1-HUVECs compared to the untreated group (Figure 4(i)). Moreover, the expression of MDA (Figure 4(m)) increased, and the expression of SOD (Figure 4(n)) decreased. Compared with the OE-Sirt1-HUVECs group, the expression of apoptosis-related protein Bax was up-regulated, and the expression of Bcl-2 was down-regulated in the HPI-4 group (Figure 4(j)–(l)). These findings suggest that CSE can inhibit the Sirt1-SHH signaling pathway, thereby causing mitochondrial dysfunction and apoptosis in HUVECs. Sirt1-SHH axis is involved in CSE-induced HUVEC apoptosis and mitochondrial dysfunction. After using SAG to activate the SHH signaling pathway: (a) MitoTracker™ Deep Red FM staining of mitochondria; (b, c) JC-1 to detect mitochondrial membrane potential; Western blot (d, e, f) assay to detect the expression of apoptosis-related proteins Bax and Bcl-2. ELISA assay to detect the expression of SOD (g) and MDA (h). After using HPI-4 to inhibit the SHH signaling pathway: (i) MitoTracker™ Deep Red FM staining of mitochondria. Western blot (j, k, l) assay to determine the expression of apoptosis-related proteins Bax and Bcl-2. ELISA assay to determine the expression of MDA (m) and SOD (n). *p < .05, **p < .01, ***p < .001.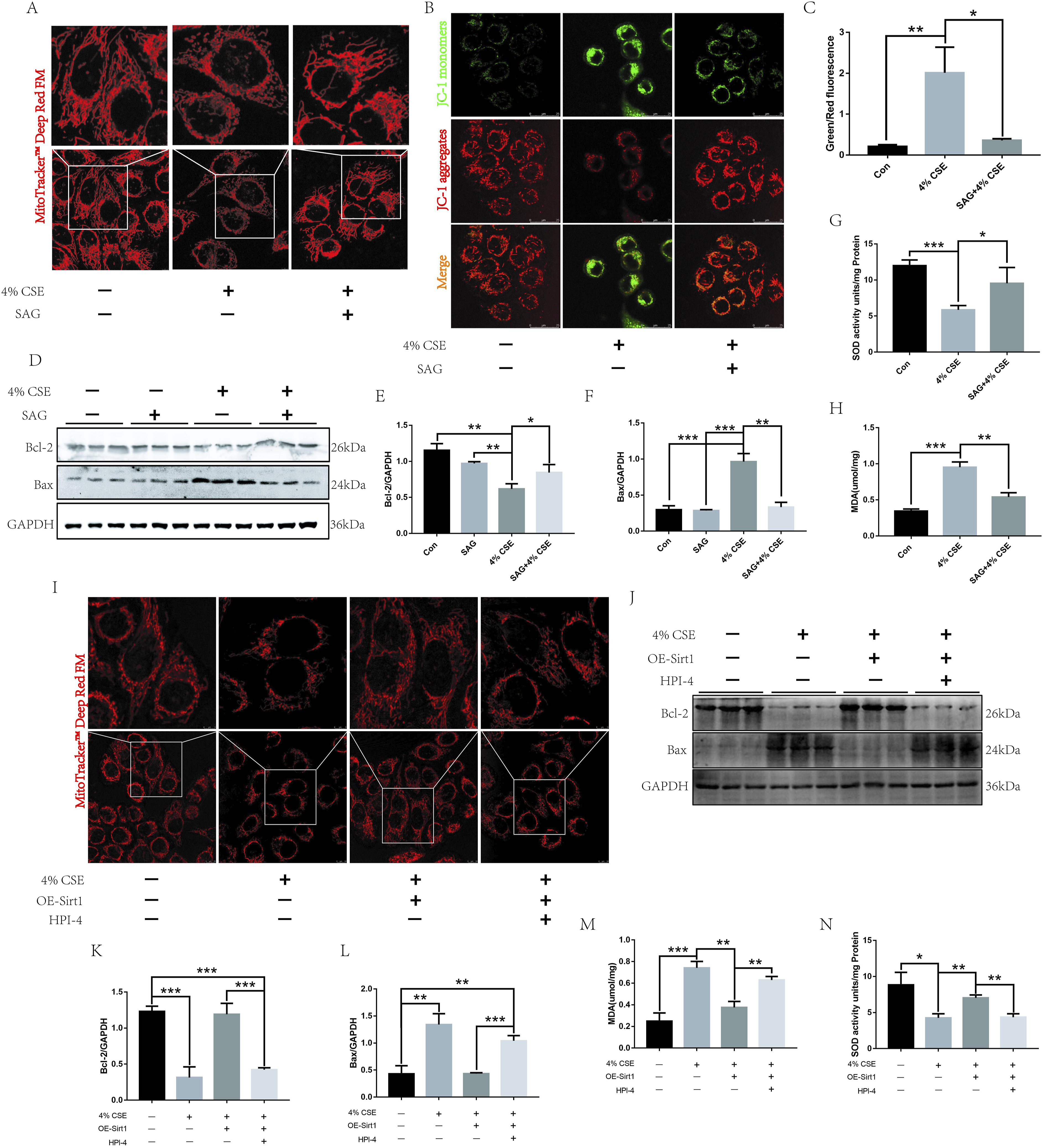
Cigarette smoke can accelerate atherosclerosis in ApoE-KO mice and inhibit the Sirt1-SHH signaling pathway
We next investigated the effect of cigarette smoke on atherosclerosis and the Sirt1-SHH signaling pathway in ApoE-KO mice. The body weights of ApoE-KO mice were monitored after the corresponding intervention (Figure 5(a)). The results showed that cigarette smoke caused weight gain (Figure 5(b)) and aggravated atherosclerosis in ApoE-KO mice (Figure 5(c)). Transmission electron microscopy showed that cigarette smoke exposure induced morphological and structural changes in aortic ECs in ApoE-KO mice compared with the control group (Figure 5(d)). Cigarette smoke also induced Bax upregulation and Bcl-2 down-regulation in the aorta of ApoE-KO mice (Figure 5(e)). Further studies also confirmed that cigarette smoke can down-regulate the expressions of Sirt1, SHH, and Gli1 in the aorta (Figure 5(f)–(h)), consistent with the results of the in vitro studies. These findings suggest that cigarette smoke may accelerate atherosclerosis and inhibit Sirt1-SHH signaling in ApoE-KO mice. Cigarette smoke can accelerate atherosclerosis in ApoE-KO mice and inhibit the Sirt1-SHH signaling pathway. (a) Administration of ApoE-KO mice; (b) Body weight of ApoE-KO mice; (c) Oil red O staining of the aorta of ApoE-KO mice; (d) Transmission electron microscopy of mouse aortic endothelial cells; (e) Western blot assay to assess the expression of apoptosis-related proteins Bax and Bcl-2 in the aorta of ApoE-KO mice; (f, g) Expression of Sirt1 detected by immunofluorescence and Western blotting; (h) Western blot assay to assess the expression of SHH and Gli1. *p < .05, **p < .01.
Discussion
Atherosclerosis is the primary pathological change underlying cardiovascular diseases.3,15 It is characterized by chronic inflammation, with innate and acquired immune cells entering the atherosclerotic plaques, significantly impacting plaque formation and stability.16,17 Accumulating experimental and clinical evidence confirms that atherosclerosis is a chronic inflammatory disease with autoimmune components.18,19
EC dysfunction is the initiating factor of atherosclerosis, which can be caused by various pathological factors (smoking, obesity, diabetes, hyperlipidemia, and hypertension). 2 Multiple studies have identified smoking as an independent risk factor for atherosclerosis. 20 Smoking can affect all stages of the atherosclerosis process, from the dysfunction of ECs to the occurrence of acute cardiovascular and cerebrovascular accidents. Both active and passive smoking can increase the morbidity and mortality of cardiovascular events. All smoking patterns (such as cigarettes, e-cigarettes, and hookah) have an impact on the occurrence of cardiovascular disease. 21 Our in vitro and in vivo studies confirmed that cigarette smoke induces EC dysfunction and accelerates atherosclerosis in ApoE-KO mice.
Smoking has long been a pressing global health concern, mainly due to its intricate pathological molecular mechanisms, which play a crucial role in inflammation and immunity.22,23 In the context of cardiovascular disease, smoking contributes to its onset and progression through multiple pathways and mechanisms. Previous studies have shown that CSE can activate the ROS/NLRP3 axis to induce apoptosis of ECs, leading to EC dysfunction. 24 Csordas et al. also showed that protein damage caused by CSE activates autophagy and eventually leads to HUVEC necrosis. Through this mechanism, smoking may lead to deterioration of vascular endothelial function, accelerating atherosclerosis. 25 The present study shows that CSE can induce apoptosis of HUVECs, leading to their dysfunction.
Sirt protein family is a class of highly conserved nicotinamide adenine dinucleotide-dependent deacetylases, including Sirt1 to Sirt7. Each member exhibits distinct tissue specificity, localization, and molecular targets, playing critical roles in various cellular processes including immune regulation, metabolism, apoptosis, and DNA damage repair.26–29 Sirt1 is also involved in regulating aging-related biological phenomena through multiple signaling pathways. 30 In the study by Yue Zhang et al., CS decreased Sirt1 to activate lung fibroblasts by promoting mitochondrial oxidative stress, which dysregulated lipid metabolism by impairing autophagy flux. 31 However, in cardiovascular diseases, Prola A et al. showed that Sirt1 can cause deacetylation of K143 on eIF2α, thereby protecting cardiomyocytes from ER stress-induced apoptosis. 32 Secondly, Winnik S et al. demonstrated a protective role of Sirt1 in atherosclerosis, mediated via activation of eNOS or reduced activity of NF-kB in ECs and macrophages. 33 Our research also suggests that Sirt1 can protect HUVECs from CSE-induced apoptosis and mitochondrial dysfunction.
Mitochondrial functional changes play an irreplaceable role in cell apoptosis. Under normal circumstances, Bcl-2 forms a heterodimer with Bax and Bak, maintaining the integrity of the outer mitochondrial membrane and preventing mitochondrial apoptosis. However, when pathological factors stimulate apoptosis, mitochondrial membrane permeability changes, leading to a decrease in mitochondrial membrane potential. This reduction in membrane potential is a hallmark indicator of apoptosis.34,35 Moreover, altered mitochondrial membrane permeability also reduces energy generation, eventually leading to cell apoptosis. Therefore, impaired mitochondrial function stability not only affects normal physiological regulation, such as development but may also induce tissue dyshomeostasis, leading to the occurrence of various diseases.36,37 Studies have shown that Sirt1 can improve mitochondrial function by alleviating inflammation and oxidative stress. 38 Previous studies have shown that Sirt1 can induce OXPHOS gene expression and mitochondrial biogenesis through deacetylation of PGC-1α. Deacetylation of PGC-1α also regulates the activation of peroxisome proliferator-activated receptor-α (PPAR-α), resulting in increased oxidation of fatty acids, thereby regulating mitochondrial function and metabolic homeostasis. 39 Similarly, in the present study, CSE was found to decrease Sirt1 expression in HUVECs. Overexpression of Sirt1 was found to significantly reduce CSE-induced apoptosis and mitochondrial dysfunction in HUVECs.
Hedgehog (Hh) signaling is evolutionarily conserved and is closely related to human development, immune regulation, and diseases. 40 While extensive research has established the role of Hh signaling pathway in development, 41 its functional maintenance in adulthood remains poorly understood. Studies have shown that deletion or mutation of the Hh genes compromises neuronal integrity and human lifespan. Conversely, activating the Hh signaling pathway has been linked to extended life span and improved health. 42 However, abnormal activation of the Hh signaling pathway can lead to the onset and progression of various tumors. 43 In the present study, CSE was found to inhibit the activation of Hh signaling pathway and decrease Sirt1 expression. However, whether Sirt1 is directly related to the Hh signaling pathway is not clear. Studies have shown that Sirt1 can stabilize Gli1 protein through deacetylation, thereby activating the Hh signaling pathway and synergically regulating the pathogenesis of myeloma drug resistance. 44 Additionally, Sirt1 has been shown to improve blood pressure, angiogenesis disorders, inflammation, and pregnancy outcomes in rats with preeclampsia by activating the SHH pathway. 45 In addition to positive regulation, Sirt1 also negatively regulates the SHH pathway. Sirt1 can inhibit the transcription of Gli1 and Gli2 proteins in the SHH pathway, thereby inhibiting the development of medulloblastoma. 46 Our study further demonstrated that CSE inhibits Sirt1 expression, leading to reduced expression of Gli1 and other related proteins in the SHH signaling pathway, thus affecting the functional changes of HUVECs.
In conclusion, our in vitro studies demonstrate that CSE induces mitochondrial dysfunction and apoptosis in HUVECs by inhibiting the Sirt1-SHH signaling pathway. Furthermore, in vivo studies using ApoE-KO mice suggest that cigarette smoke accelerates atherosclerosis through this mechanism. These findings provide new insights into the prevention and treatment of smoking-induced atherosclerosis.
Footnotes
Statements and Declarations
Author contributions
All authors contributed to this research paper. Weiming Wang, Gang Yuan, Guang Li designed and completed the experiments, as well as drafted and wrote the manuscript. Youhua Xu, Yue Chen, and Tingting Zhao edited and provided critical review of the manuscript. All authors discussed and confirmed the final manuscript.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was funded by the Luzhou Science and Technology Plan Project (No. 2021-JYJ-57), Open Program of Nuclear Medicine and Molecular Imaging Key Laboratory of Sichuan Province (No. HYX22002), Doctoral Research Initiation Fund of Affiliated Hospital of Southwest Medical University, Southwest Medical University campus level research project (No. 2022QN070), and Open Fund of the Key Laboratory of Medical Electrophysiology of Ministry of Education and Sichuan Province (No. KeyME-2020-008).
Conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Data Availability Statement
The datasets used and analyzed during the current study are included in this published article.