
Abstract
Endothelial damage plays a key role in atherosclerosis and this is impacted upon by numerous risk factors including cigarette smoking. A potential measure to reduce the cardiovascular burden associated with smoking is to reduce smoke toxicant exposure. In an in vitro endothelial damage repair assay, endothelial cell migration was inhibited by cigarette smoke particulate matter (PM) generated from several cigarette types. This inhibition was reduced when cells were exposed to PM from an experimental cigarette with reduced smoke toxicant levels. As a number of toxicants induce oxidative stress and since oxidative stress may link cigarette smoke and endothelial damage, we hypothesized that PM effects were dependent on elevated cellular oxidants. However, although PM-induced cellular oxidant production could be inhibited by ascorbic acid or n-acetylcysteine, both these antioxidants were without effect on migration responses to PM. Furthermore, reactive oxygen species production, as indicated by dihydroethidium fluorescence, was not different in cells exposed to smoke from cigarettes with different toxicant levels. In summary, our data demonstrate that a cardiovascular disease–related biological response may be modified when cells are exposed to smoke containing different levels of toxicants. This appeared independent of the induction of oxidative stress.
Keywords
Introduction
Atherosclerotic cardiovascular disease (CVD) is a major cause of mortality in the Western world. The defining characteristic of this disease is the formation of atherosclerotic lesions or plaques which occlude blood vessels and disrupt blood flow. This leads to acute manifestations such as myocardial infarction and stroke in which tissue oxygen and nutrient supply are severely compromised. A complication of these plaques is their vulnerability to rupture, giving rise to a thrombus with the ability to occlude vessels away from the initial plaque site. 1 –3 Although atherosclerosis was initially considered a simple disease involving arterial lipid accumulation, it is now known to involve a cascade of inflammatory processes. 1 –3 The initiating step in the development of an atherosclerotic lesion is damage to the endothelium. 4,5 The endothelium is composed of a monolayer of endothelial cells lining blood vessels and acts as a master regulator of vascular function. In a healthy individual and prior to the onset of CVD, the endothelium plays a homeostatic role in maintaining vascular tone and blood flow. 4 In the early stages of CVD development, endothelial damage and dysfunction triggers a chronic inflammatory process in the vessel wall. This ultimately involves numerous other cell types including vascular smooth muscle cells, monocytes (which become tissue macrophages and subsequently foam cells), and platelets. 6 –8
As mentioned above endothelial damage and dysfunction is a critical initiating step in atherosclerosis 4,5,7,9 as well as in initiating pathogenesis resulting from surgical procedures such as balloon angioplasty. 10 –12 While endothelial injury itself initiates atherosclerosis, endothelial repair by the processes of migration and proliferation restores endothelial integrity and is atheroprotective. 3,13 –16 For example, viral overexpression of the vascular endothelial growth factor (VEGF), a stimulant of migration and proliferation, accelerated endothelial repair and inhibited neointima formation following arterial injury. 17 In the same article, it was reported that sequestration of VEGF delayed the regrowth of the endothelial layer and increased neointima size. Similar findings had previously been reported in a balloon-injured rat carotid artery model, 18 while a more recent study in an atherosclerosis-prone mouse model demonstrated that impairment of re-endothelialization caused enhanced neointima formation. 16 The latter study showed that the mechanism underlying this response was impaired endothelial proliferation and migration.
Smoking is recognized to cause gross structural damage to the endothelium and endothelial dysfunction. 19 –23 This may, at least in part, explain why smoking is a risk factor strongly associated with the development of CVD. In 2001, the US Institute of Medicine (IOM) reported that, since smoking-related diseases were dose related and because epidemiological studies showed a reduction in the risk of smoking-related diseases following cessation, it might be possible to reduce smoking-related risks by developing potential reduced-exposure products (PREPs). These were defined as products that 1 result in the substantial reduction in exposure to one or more tobacco toxicants and, 2 if a risk reduction claim is made, products that can reasonably be expected to reduce the risk of one or more specific diseases or other adverse health effects. 24 More recently the IOM published, after a request from the US Food and Drug Administration, a further report that focused on scientific studies to assess modified risk tobacco products. 25 These reports have proposed that, alongside other forms of data (from clinical studies for example), data from disease-relevant in vitro models are pivotal to the assessment of the biological effects of reduced toxicant cigarettes. Data from all of these sources may contribute to product evaluation. 24,25
In this study, we describe the application of an in vitro endothelial damage repair (scratch wound) assay which is responsive to cigarette smoke extracts and demonstrate that modification of machine-measured particulate phase smoke toxicant yields reduced the inhibitory effects of these extracts on endothelial damage repair. Our data support the suggestion that in vitro models of smoking-related diseases may be of value, as part of a framework of tests, in assessing the biological effects of smoke from cigarettes with different yields of smoke toxicants.
Materials and Methods
Materials
Endothelial basal medium (EBM-2) was supplied by Lonza (Basel, Switzerland). This medium was supplemented by the addition of human epidermal growth factor, hydrocortisone, gentamicin, amphotericin-B, 2% fetal bovine serum (FBS), VEGF, human fibroblast growth factor-B, R 3 -insulin-like growth factor 1, heparin, and ascorbic acid. These supplements were all part of the SingleQuot kit (Lonza). ReagentPack subculture reagent kit containing (4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid) (HEPES)-buffered saline, trypsin/EDTA, and trypsin neutralizing solution was from Lonza. Ascorbic acid, n-acetylcysteine (NAC), and neutral red dye solution were from Sigma (Poole, UK). Dihydroethidium was supplied by Invitrogen (Paisley, Scotland). ImageLock plates were from Essen Instruments (Ann Arbor, Michigan). 3R4F reference cigarettes were obtained from the University of Kentucky.
Generation of Cigarette Smoke Particulate Matter
Cigarette smoking causes cardiovascular diseases and particulate matter ([PM], portion of the whole cigarette smoke) contributes to this association. 26 All tests were performed on PM trapped on a Cambridge filter pad after smoking experimental cigarettes using a RM20 smoking machine (Borgwaldt-KC, Hamburg, Germany), according to the parameters outlined in International Organization for Standardization (ISO) 3308 (2000). Depending on the machine-smoked particulate delivery of a given cigarette, an appropriate number of cigarettes were smoked to obtain approximately 300 mg of PM on a 44-mm Cambridge filter pad (Whatman, Maidstone, UK). Typically, this was in the range of 30 to 60 cigarettes per pad, which is more than is typically collected in standardized testing regimes aimed at measuring PM. After smoking, the Cambridge filter pads were cut into 2 segments with the large segment being 3 quarters of the total pad. After weighing to determine the weight of the particulate deposited, the larger segment was immediately extracted into dimethylsulfoxide (DMSO) to a concentration of 24 mg/mL. The remaining quarter of the Cambridge filter was used to confirm nicotine-free dry PM, water and, where applicable, glycerol yields. Particulate solutions were stored, protected from light in single-use aliquots at −20°C for no longer than 1 month, which preserves the stability of the PM (data not shown). The stock PM solution was defrosted prior to each experiment and dilutions of this stock were made in media to the required experimental concentrations. Particulate matter solutions are stable under normal light conditions over the time period of the setting up of an experiment. All of the assays themselves were performed in the dark inside incubators which ensured that the PM remained stable.
In the majority of experiments, PM was obtained from 3R4F reference cigarettes (University of Kentucky College of Agriculture; http://www.ca.uky.edu/refcig/3R4F%20Preliminary%20Analysis.pdf). To examine the effects of cigarette smoke toxicant reduction on biological responses, 2 test cigarettes were manufactured for these studies. One 6 mg ISO tar product was manufactured as a conventional control cigarette (coded CC6). This was based on British American Tobacco cigarettes on sale in Europe at the time of the studies and contained 100% US blend tobacco. A 6 mg ISO tar yield reduced toxicant prototype cigarette (coded TSS6) was constructed using a blend of 80% US blend tobaccos and 20% tobacco substitute sheet material, and a 2-segment filter containing 80 mg of polymer-derived carbon. Details of the designs and the associated smoke chemistries have been reported elsewhere (ref 27 and see Supplementary Table 1).
Cell Culture
All experiments were carried out on human umbilical vein endothelial cells (HUVECs) obtained from either Lonza or Lifeline Cell Technology (Walkersville, Maryland). These cells were cultured in a 5% CO2 (balance air) humidified atmosphere at 37°C in supplemented EBM-2 media (see Materials section). Cells were passaged twice-weekly by trypsinization, centrifugation, and resuspension and seeded in new vessels. Cells were only used up to passage 5.
Endothelial Scratch Wound (Migration) Assay
The HUVECs were seeded in 24-well (6 × 4) ImageLock plates 24 to 48 hours prior to experimentation to allow them to grow to confluency. On the experimental day, confluency was checked by carrying out a single scan of each well in the IncuCyte imaging apparatus (Essen Instruments). Only when all wells were fully confluent were experiments performed. After confirming full confluency, culture media was changed from EBM-2 media containing 2% FBS to EBM-2 containing a nominal amount of FBS (0.1%). This inhibits cell proliferation and allows the examination of endothelial migration in isolation. Six hours after this media change, a scratch wound was made in each well using the Essen Woundmaker apparatus. Four sterile 10-μL pipette tips were attached to the Woundmaker and lowered into 4 wells of the ImageLock plate. The plate was moved backward and forward 3 times to create scratch wounds in these wells. This procedure was repeated for a further 2 columns of 4 wells before the tips were changed to scratch a further 3 columns. After wounding, test agents (PM or positive control agents) were added to the wells of the ImageLock plates and these were then placed inside the IncuCyte imaging apparatus. Images of the cells taken by the IncuCyte apparatus were checked visually to ensure that clean scratch wounds had been created in each well. Subsequently, 3 individual images of each scratch wound were taken at hourly intervals over a period of 20 hours. Wound width was measured using the IncuCyte software (Essen Instruments).
Effects of PM Derived From Cigarettes With Altered Smoke Toxicants
The migration assay was used to examine the effects of PM generated from an experimental cigarette with altered smoke chemistry due to the inclusion in the cigarette of a tobacco substitute sheet (TSS6 27,28 ). The effects of this PM were compared to those seen in cells generated from a conventional control cigarette with matched tar content (CC6). When examining differences between the control and the modified cigarette, each comparison was made by performing scratch assays at least twice at different cell passage numbers (passages 4 and 5) in each experiment, with 4 experimental repeats in each case. In a given experiment, direct comparisons were made between control and test cigarettes examined in the same ImageLock plates.
Vascular Endothelial Growth Factor Removal
To examine the effects of removing the VEGF on endothelial migration, cells were seeded as described above. On the experimental day, after determining confluency the culture media was changed for one containing 0.1% FBS and no VEGF. Six hours later, scratch wounds were made as described previously and fresh media containing 0.1% FBS and no VEGF was placed into the wells.
Neutral Red Uptake (Cell Viability) Assay
Cells were exposed to various concentrations of PM in growth media for 24 hours. The cultures were then incubated with a 1:65 (v/v) neutral red dye solution in media for 3 hours at 37°C to allow active uptake of the dye into viable cells. Cells were washed twice with phosphate-buffered saline to remove unincorporated dye. The neutral red solution was eluted from the cells by the addition of 200 μL of distilled water containing 50% ethanol and 1% acetic acid, followed by gentle agitation for 20 minutes. The viability of the cultures was determined by measuring the active uptake of neutral red dye into live cells using a microplate reader (Thermo Labsystems, Waltham, Massachusetts) at an excitation wavelength of 540 nm and an emission wavelength of 630 nm. Blank inserts were included to establish background levels of dye that were absorbed onto the insert matrix in the absence of cells. These background measurements were subtracted from those of the treated and untreated control cultures.
Monitoring the Intracellular Formation of Free Radicals
All experiments were conducted under low light conditions as the superoxide reported dye, dihydroethidium (DHE), is light sensitive. The fluorescent probe was prepared freshly before use on each experimental occasion. A 2.5 mmol/L stock solution of DHE in DMSO was diluted to 2.5 μmol/L in HEPES physiological saline solution, which was composed of (in mmol/L) NaCl (137), KCl (4.7), HEPES (10), NaH2PO4 (1), MgCl2 (0.56),
Fixing and Mounting
Cells were fixed with 4% formaldehyde for 25 minutes at room temperature and washed twice with HEPES solution. After fixing, coverslips were mounted onto a glass microscope slide with fluorescence mounting medium (Sigma). The edges of the coverslip were dried with blotting paper and then sealed with clear nail varnish.
Imaging and Image Analyses
After fixing, samples were imaged immediately using an inverted fluorescence microscope (Nikon, Surrey, UK) equipped with an Orca 12-bit camera (Hamamatsu, Japan) coupled to IPLAB version 4 imaging software. Cells were excited (510-560 nm) and emitted fluorescence (573-648 nm) monitored using an appropriate filter set (Chroma, Rockingham, Vermont). Fluorescence levels were quantified using ImageJ version 1.41 (National Institutes of Health, Bethesda, Maryland). To quantify fluorescence, line scan pixel intensity analysis was performed on 5 randomly selected cells in each field of view. The peak fluorescence along this line was normalized to the background intensity and averaged for the 5 cells.
Antioxidant Treatments
In experiments using ascorbic acid and NAC, cells were loaded with these antioxidants for 5 hours prior to either performing the scratch wound assay or treating cells with PM before measuring fluorescence. Antioxidants were also present for the period of exposure of the cells to PM.
Statistics
Data are presented as means (± standard error of the mean [SEM]). Comparisons were made using unpaired Student t tests. Wound width data from migration assays are presented as a percentage of the wound width at the start of the experiment. Data were averaged from the numbers of individual scratch wounds stated and in at least 2 independent experiments. To determine rate constants for endothelial migration, wound width data were analyzed using a kinetic approach. Both zero- and first-order kinetics very closely described this migration. In imaging studies, image intensities were averaged from 5 independent fields of view and from 5 randomly selected cells in each field of view.
Results
Cigarette Smoke Extracts Inhibit Endothelial Migration
In an endothelial scratch wound assay and as shown in Figure 1A, under control conditions endothelial cells migrated into the wound and caused a reduction in wound width. This migration was VEGF dependent since removal of VEGF from the culture media prior to and following wounding inhibited this reduction in wound width (Figure 1B). Thus, the wound width (expressed as a percentage of the wound width at the start of the experiment) 20 hours following wounding was 40.7% ± 2.7% (n = 45) in the presence of VEGF and 65.0% ± 3.1% (n = 45) in the absence of VEGF (P < .05, Student unpaired t test). Endothelial migration was also inhibited by 0.5 to 1 μg/mL cytochalasin D and 0.1 to 1 μg/mL endostatin (data not shown).
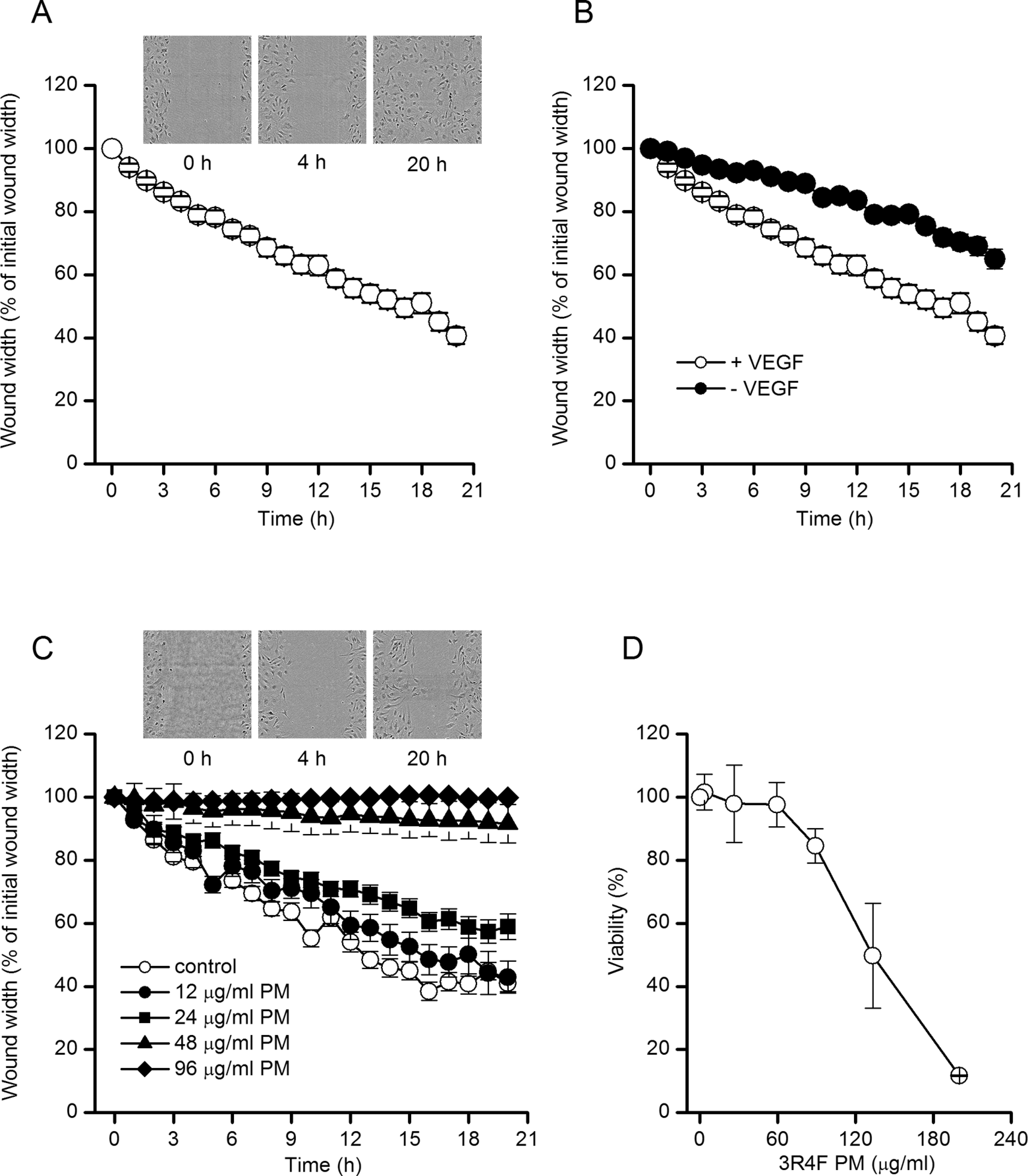
Cigarette smoke extracts inhibited endothelial migration. A, Time series plot showing the in vitro migration of HUVECs. Data are means ± SEM n = 45). Images above the plot are exemplar images of a scratch wound from the indicated time points after wounding. B, As in A, except the cells were cultured in media in the presence (•) or absence (^) of the vascular endothelial growth factor (VEGF; n = 45 in each case). C, The PM from 3R4F reference cigarettes inhibited endothelial migration in a concentration-dependent manner. Images above the plot are exemplar images of a scratch wound from the indicated time points after wounding in the presence of 24 μg/mL 3R4F PM. In panels A to C, data were normalized to the initial wound width immediately after wounding. Data are means (± SEM) between 10 and 31 scratch wounds in at least 2 independent experiments in each case. D, cell viability, determined by neutral red uptake assays, in HUVECs exposed to 3R4F PM. Data are means ± SEM from 3 experiments at each concentration. HUVECs indicates human umbilical vein endothelial cells; PM, particulate matter; SEM, standard error of the mean; VEGF, vascular endothelial growth factor.
We then examined the effects of cigarette smoke PM on endothelial migration. In these studies, PM was applied to cells immediately after creating the scratch wound. Images of the wound were taken every hour for a period of 20 hours and wound width measured using IncuCyte software. In the presence of PM from 3R4F reference cigarettes, endothelial migration was impaired such that the smoke extract caused a concentration-dependent decrease in wound recovery (Figure 1C). The concentration dependency of this response was steep, such that a concentration of 12 μg/mL of 3R4F PM was without statistically significant effect on endothelial migration. Concentrations higher than this, however, caused a reduction in endothelial migration. For example, 20 hours after wounding the wound width (expressed as a percentage of the wound width at the start of the experiment) was 40.7% ± 2.7% (n = 45) in control cells. In cells exposed to 24, 48, and 96 μg/mL of 3R4F PM, this was significantly increased to 59.0% ± 4.0% (n = 21), 91.6% ± 6.1% (n = 31), and 99.9% ± 0.4% (n = 25), respectively (P < .05 compared to controls in each case, Student unpaired t test). Neutral red uptake assays (Figure 1D) demonstrated that at 3R4F PM concentrations of 24 and 48 μg/mL, there was no significant reduction in cell viability even though a reduction in endothelial migration was observed. However, at the 3R4F PM concentration of 96 μg/mL, the inhibition of endothelial migration was accompanied by a reduction in endothelial viability (Figure 1D). In control studies, the solvent DMSO was found to have no effect on endothelial migration at concentrations to which cells were exposed in the presence of 3R4F PM (data not shown).
Smoke Toxicant Alteration Mitigates the Effects of PM on Endothelial Migration
Modification of cigarette smoke toxicant levels has been proposed as a means by which to reduce the harmful effects of smoking. 24,25 Here, we examined the effects on endothelial migration of PM derived from an experimental cigarette that had yields of a number of smoke toxicants which were lower than those of conventional control and 3R4F cigarettes (28 and Supplementary Table 1). As illustrated in Figure 2, exposure of cells to PM derived from the experimental (TSS6) cigarette had a lesser effect on endothelial migration when compared to PM from the matched control (CC6) cigarette. For example, wound width (expressed as a percentage of the original wound width) 20 hours after wounding and exposing cells to 48 μg/mL PM was 73.7% ± 3.4% when using PM from the CC6 cigarette, and this was significantly reduced to 57.8% ± 3.5% when using PM from the TSS6 cigarette with altered smoke toxicants (n = 12 in each case; P < .05, unpaired Student t test). When assessing the same parameter, the effects of 3R4F PM were significantly greater than those of TSS6 PM at a concentration of 24 μg/mL (Figure 2C). Although this effect was insignificant at a PM concentration of 48 μg/mL (Figure 2C), these data further demonstrate that PM derived from cigarettes with different toxicant yields may exert different effects on endothelial migration in this assay.
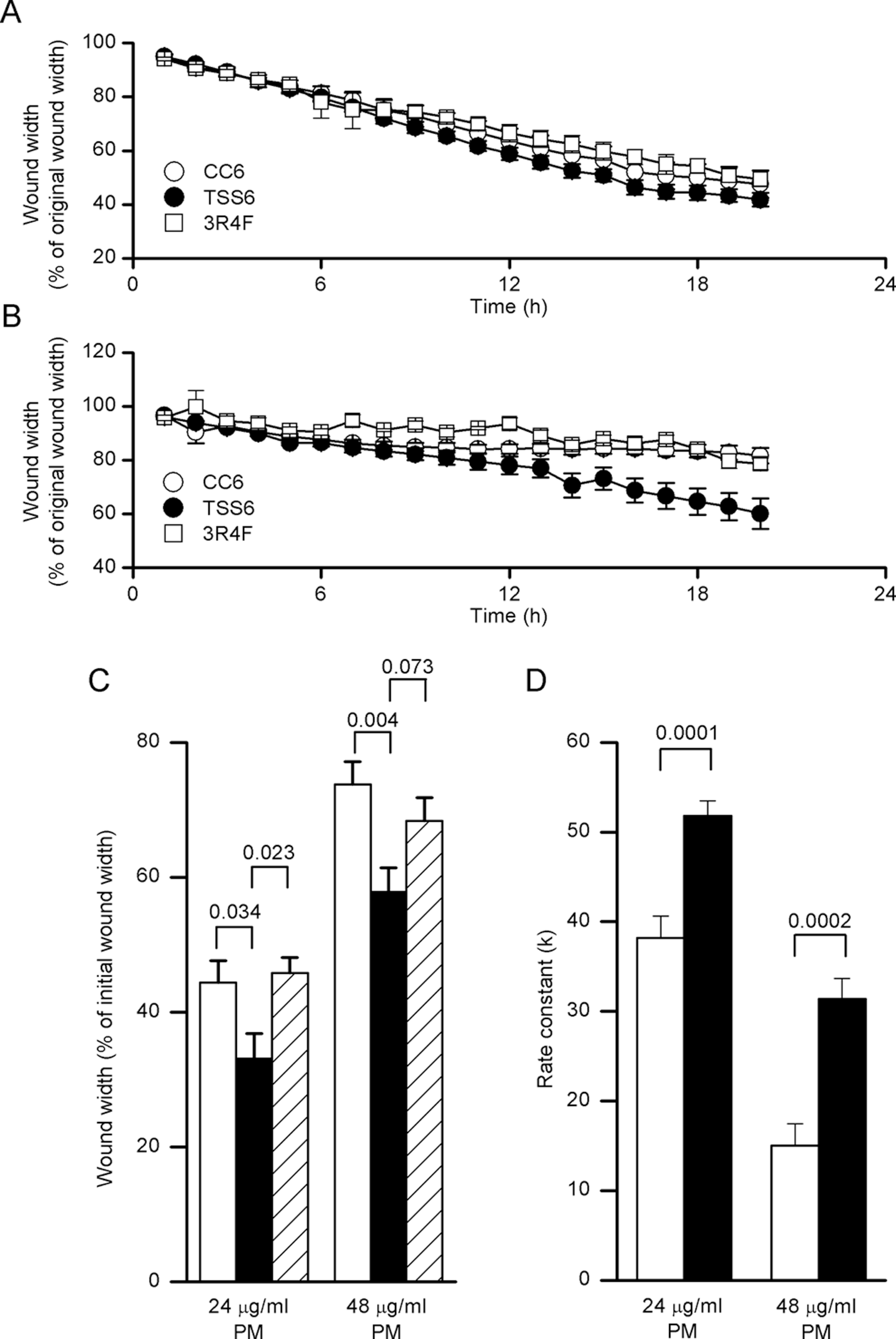
Smoke toxicant alteration mitigates the effects of PM on endothelial migration. A, Representative time series plot showing the in vitro migration of HUVECs. Cells were exposed to 24 μg/mL PM from either a conventional control (CC6) cigarette, a cigarette with altered levels of smoke toxicants (TSS6) or the 3R4F reference cigarette, as indicated. B, As in A, except the cells were exposed to 48 μg/mL of PM. C, Wound width in scratch assays 20 hours after the cells were exposed to PM at the indicated concentrations. D, Endothelial cell migration rates in cells exposed to PM from CC6 and TSS6 cigarettes. Zero-order rate constants (k′) were normalized to the nicotine levels in smoke from CC6 (0.49 mg) and TSS6 (0.43 mg) cigarettes. In C and D, PM was derived from CC6 (open bars), TSS6 (filled bars), or 3R4F (hashed bars) cigarettes. Numbers above bars are P values (Student unpaired t tests) and data are means (±SEM) averaged from 2 to 3 independent experiments with 4 experimental repeats in each case. HUVECs indicates human umbilical vein endothelial cells; PM, particulate matter; SEM, standard error of the mean.
Nicotine levels in the smoke from CC6 and TSS6 cigarettes are moderately different (see Supplementary Table 1). Given this difference, we examined whether nicotine contributed to the difference in effects on endothelial migration seen when exposing cells to PM from these cigarettes by normalizing migration data to the cigarette smoke nicotine content. In this analysis, wound closure data were analyzed using a kinetic approach. Both zero- and first-order kinetics very closely described the endothelial migration; data reported here are for zero-order rate constants. For both CC6 and TSS6, the rate constants for the higher (48 μg/mL) PM concentrations were smaller, indicating slower wound closure at higher smoke particulate concentrations (Figure 2D). In contrast, the rate constants for the experiments involving PM from TSS6 were higher than those for CC6 cigarette smoke PM, reflecting quicker wound closure from the modified cigarette smoke condensate. This was significant at both 24 and 48 μg/mL PM (Figure 2D). Thus, the different nicotine contents of the cigarette smoke cannot explain the difference in effects on endothelial migration. In support of this, although the smoke from 3R4F and CC6 cigarettes exhibited markedly different nicotine levels (0.7 and 0.49 mg/cigarette, respectively), cells exposed to smoke from these migrated at similar rates (Figure 2B).
Reactive Oxygen Species Production in Endothelial Cells Exposed to PM
Previous studies have suggested that elevated reactive oxygen species (ROS) levels may regulate endothelial migration. 29 –31 Furthermore, a number of cigarette smoke toxicants that exist in the particulate fraction of the cigarette smoke, for example benzo(a)pyrene, have been linked to the production of cellular ROS and the induction of oxidative stress. 32,33 Initially, to determine the ability of cigarette smoke extracts to induce the formation of ROS we performed imaging studies in cells treated with 3R4F PM and subsequently loaded with the superoxide reporter dye, DHE. After a 24-hour treatment with 3R4F PM, cellular fluorescence was increased in a concentration-dependent manner, indicative of elevated ROS production (Figure 3A). This elevation of fluorescence was abolished when cells were exposed to PM following a 5-hour preincubation with, and in the presence of, the antioxidants ascorbic acid (200 μmol/L) or NAC (5 mmol/L; Figure 3B). Neither antioxidant had any effect on DHE fluorescence levels in the absence of PM, indicating that the effect of the antioxidants was to abolish the increase in fluorescence due to PM exposure and not due to a nonspecific effect (Figure 3B). Thus, PM induced an antioxidant-sensitive elevation of superoxide levels in HUVECs. The elevation in the cellular levels of this form of ROS was not different between any of the 3 cigarette types used in these studies. Thus, DHE fluorescence (normalized to the fluorescence observed in cells exposed to PM from 3R4F reference cigarettes) was not significantly different in cells exposed to PM from either the CC6 or TSS6 cigarettes (Figure 3C).
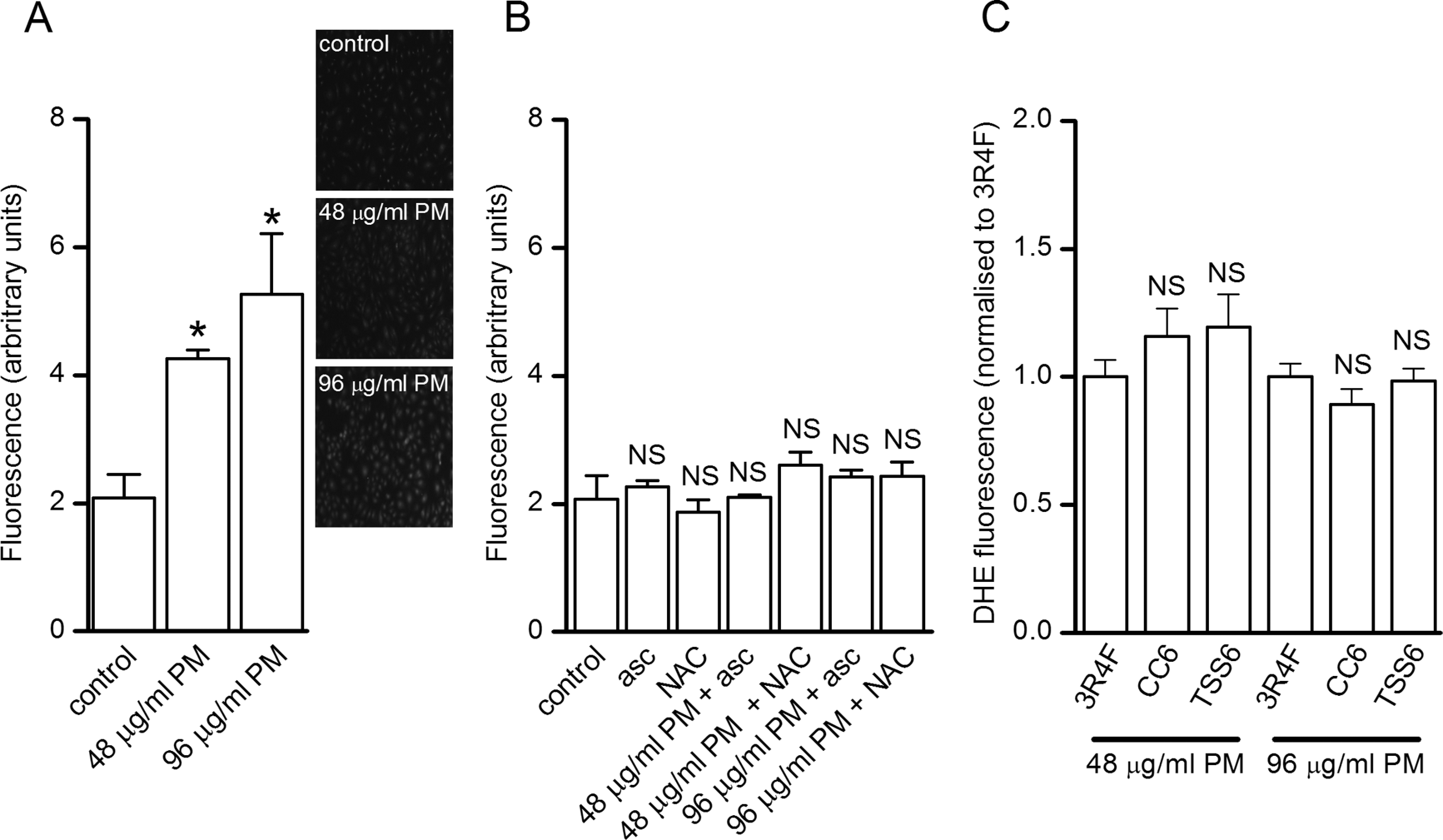
Cigarette smoke PM induced ROS production in HUVECs. A, In cells loaded with the ROS-sensitive dye DHE, 24-hour treatment with PM from 3R4F reference cigarettes induced a concentration-dependent increase in fluorescence. B, Antioxidant treatment abolished the fluorescence increase due to 3R4F PM. Asc, 200 μmol/L ascorbic acid; NAC, 5 mmol/L n-acetylcysteine. C, Effects of 24-hour treatment with 3R4F, CC6, and TCC6 PM on fluorescence in HUVECs loaded with DHE. Data were normalized to fluorescence values in cells treated with PM from the 3R4F reference cigarette. In all panels, image intensities were averaged from 5 independent fields of view and from 5 randomly selected cells in each field of view and data are mean (±SEM) values. *P < .05 compared to control. NS, not significantly different compared to control (B) or 3R4F (C). DHE indicates dihydroethidium; HUVECs, human umbilical vein endothelial cells; PM, particulate matter; ROS, reactive oxygen species; SEM, standard error of the mean.
Antioxidants Were Without Effect on PM-Induced Inhibition of Endothelial Migration
To explore our hypothesized link between PM exposure, ROS production and inhibition of endothelial migration, endothelial cells were preincubated with antioxidants for 5 hours prior to wounding and also during the exposure to 3R4F PM following wounding. These conditions were identical to those described above in the studies measuring ROS levels with DHE. As shown in Figure 4, inhibition of endothelial migration in response to both 24 and 48 μg/mL 3R4F PM was unaffected by either 200 μmol/L ascorbic acid (Figure 4A) or 5 mmol/L NAC (Figure 4B). These concentrations of antioxidants were effective in inhibiting the PM-induced elevation of ROS levels in HUVECs (see Figure 3). Thus, in our studies antioxidants had no effect on 3R4F PM-induced inhibition of endothelial migration and, therefore, it is likely that ROS do not play a role in this inhibitory response to PM. This lack of a role for ROS is supported by the data presented in Figures 2 and 3C which showed that migration rates were different in cells exposed to PM from different cigarettes but that the elevation of ROS in cells exposed to these PM were not significantly different.
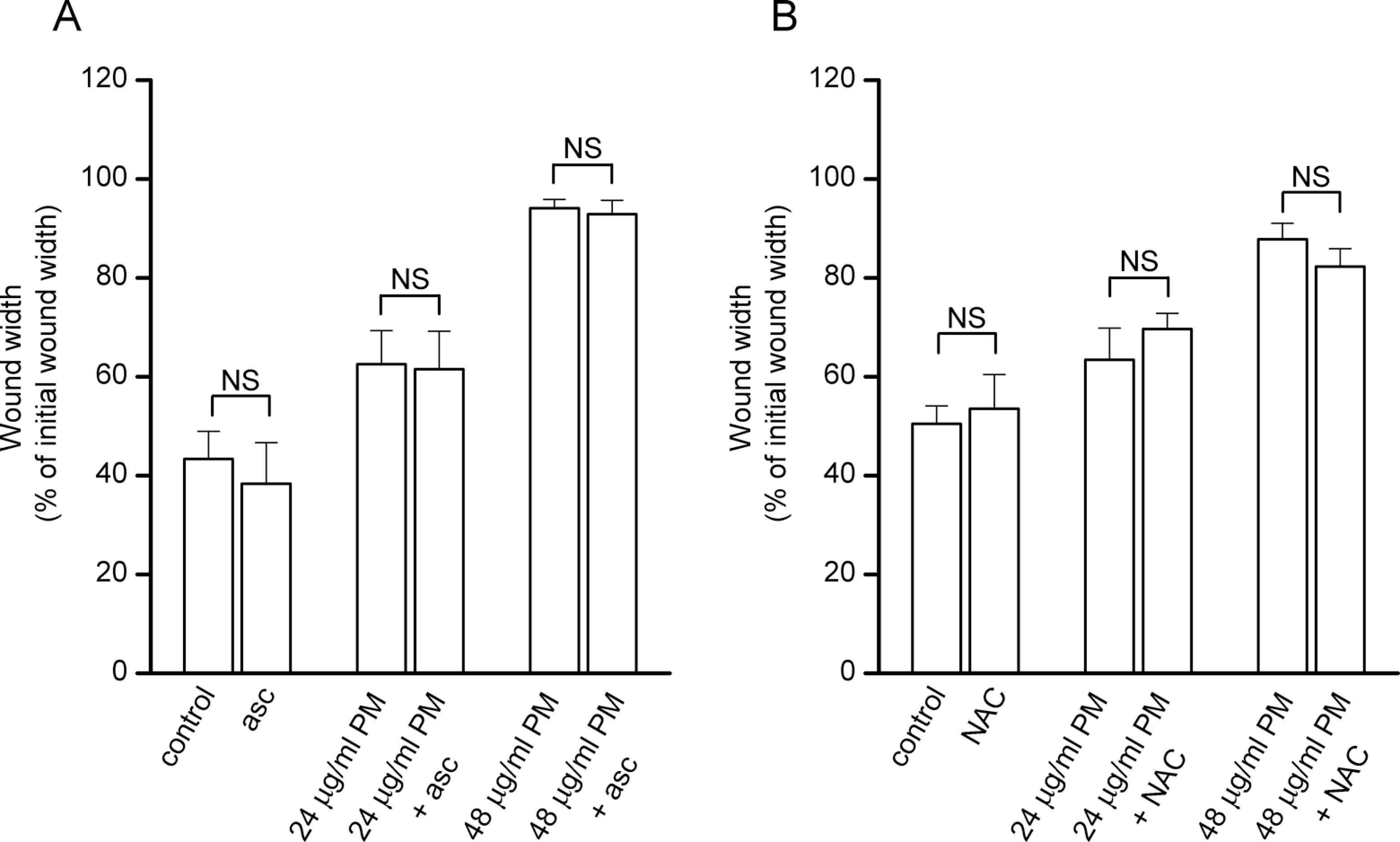
Antioxidant treatment was without effect on PM-induced inhibition of endothelial migration in vitro. Mean (±SEM) wound widths in HUVECs treated with the indicated combinations of PM from 3R4F reference cigarettes and either (A) ascorbic acid (asc, 200 μmol/L) or (B) n-acetyl-cysteine (NAC, 5 mmol/L). Wound widths were measured from images taken 20 hours after wounding cells. In A and B, data were taken from between 7 and 16 scratch wounds in each case and from at least 2 independent experiments. NS, not significantly different compared to wound width in experiments performed in the absence of the antioxidant (Student unpaired t test). HUVECs indicates human umbilical vein endothelial cells; NAC, n-acetylcysteine; PM, particulate matter; SEM, standard error of the mean.
Discussion
Cigarette smoking has long been associated with CVD and this link has been strengthened by many studies examining the effects of cigarette smoke extracts in disease-relevant in vitro model systems utilizing cardiovascular system cells. Cigarette smoke is a complex and dynamic mixture of more than 5600 identified chemical constituents 34 in both its particulate and vapor phases. Components of each of these phases may contribute to the pathogenic effects of cigarette smoking. In 2001, the IOM reported that, since smoking-related diseases were dose related, and because epidemiologic studies show a reduction in the risk of smoking-related diseases following cessation, it might be possible to reduce smoking-related risks by developing PREPs which would result in the substantial reduction in exposure to one or more tobacco toxicants. 24 Despite this proposal however, information concerning which of the many toxicants is most crucial to reducing the CVD risk is limited.
Cigarette smoke toxicant reductions may be achieved by numerous different approaches. 28 Blend modification, tobacco treatment and selective toxicant filtering are some of the ways in which cigarette smoke toxicant yields may be reduced. 35 Once a reduced toxicant cigarette is generated, it must be evaluated in terms of assessing exposure and toxicity, individual health risk, and population harm. 35 Key components of such evaluations are preclinical (nonhuman) studies using in vitro toxicological tests, and these may provide supporting evidence, alongside smoke constituent yield analysis, in vivo animal studies, premarket human studies and postmarket testing, of modified individual and population risk. 24,25,35 Prior to our studies described here however, there have been no reports of studies demonstrating that altering smoke toxicant yields modifies end points in CVD in vitro models.
Endothelial damage is a key initiating step in CVD, and it is currently thought that a potential mechanism underlying the effects of cigarette smoking in enhancing CVD risk may be explained, at least in part, by impaired endothelial repair processes in smokers. 15 In this study, we describe an in vitro CVD model in which endothelial cells in culture were damaged. Visual imaging of the cells allowed the measurement of the inhibitory effects of cigarette smoke PM on endothelial migration. The effects of PM in this model were dose dependent and occurred independently of any effects on cell viability. Using this model, we examined the effects of PM derived from a cigarette which due to blend and filter modifications had reductions in the machine yields of a number of cigarette smoke toxicants. The inhibitory effects on endothelial migration of PM derived from this cigarette were significantly lower than those seen when using PM from a matched control (conventional) cigarette and also from the 3R4F reference cigarette, although only at the lower concentration used for the latter. In our previous studies in which the effects of smoke from cigarettes modified by the inclusion of glycerol-containing tobacco substitute sheet were examined in several bacterial and mammalian in vitro cytotoxicity assays, such modification reduced smoke cytotoxicity. 27 That is, cytotoxicity was greater for the control cigarette smoke when examined by comparing toxicity at matching exposures to PM. However, in each assay when cytotoxicity was examined by comparing exposures on the basis of exposure to nicotine- and humectant-free dry PM (ie, allowing for the glycerol content of the PM), there were no significant differences in toxicity. This suggests that the reduction in cytotoxicity was related to the dilution of the cigarette smoke by the inclusion of the tobacco substitute sheet. Although we are unable to perform such an analysis on the data presented in this article, potentially the reduced effects of the cigarette smoke from the modified cigarettes on endothelial migration may be accounted for by the glycerol dilution.
Previous studies have documented modified responses of biological end points (mutagenicity, genotoxicity, and cytotoxicity) in in vitro assays to smoke and/or extracts from modified cigarettes. 36 –38 To our knowledge however, our data are the first to describe a modified response in an in vitro model of a smoking-related disease to smoke from a modified cigarette and this has 2 major implications. First, our data suggest that in vitro models are indeed a means of examining the effects of smoke from modified cigarettes and as described above such models may, therefore, be used in the future as product assessment tools for reduced toxicant cigarettes. Due to the complex nature of atherosclerotic CVD, it may not necessarily be possible to directly extrapolate data from studies using in vitro models to estimate human health risk. The models may however provide important and necessary supporting information of modified risk, as part of a weight-of-evidence approach such as that described above examining the end points in both preclinical and clinical studies. Second, our data support the notion that altering cigarette smoke toxicant levels may modify biological responses to cigarette smoke. However, further studies are required to examine the human exposure to toxicants from modified cigarettes and also to define the clinical implications of reducing smoke toxicant levels, which is necessary to provide a stronger weight-of-evidence for any reduced risk potential.
An outcome of the use of the model described in the current study is an examination of the potential mechanisms underlying the response of the migration assay to PM. Constituents of the vapor and particulate phases of cigarette smoke are both sources and inducers of cellular free radical production, and it is recognized that cigarette smoke can induce the generation of oxidative species by cellular enzyme systems, while also decreasing antioxidant protection. 39,40 Of further interest, ROS may regulate endothelial migration, 29 –31,41 and this potentially provides a link between smoking and atherogenesis. Our findings that cigarette smoke PM induced free radical production in endothelial cells are in accordance with those reported previously, in which potential sources of free radicals have been suggested as either the plasmalemmal nicotinamide adenine dinucleotide phosphate (NAD(P)H) oxidase, 41,42 the mitochondria 43 or xanthine oxidase. 44 However, the apparent lack of a role for elevated ROS levels in mediating the effects of PM on endothelial migration in our studies contrasts with the previous findings demonstrating that smoke extract–mediated inhibition of VEGF-induced HUVEC migration was reversed by either NAC or vitamin C (ascorbic acid). 29 In the studies conducted by Michaud et al, 29 cigarette smoke extracts were produced by bubbling cigarette smoke through tissue culture media, which captures water-soluble components of both the vapor and particulate phases. In our studies however, extracts were produced by capturing cigarette smoke on a Cambridge filter pad and eluting the trapped particulate using DMSO. This experimental difference has the potential to explain these differential findings and leads to the possibility that components of the different phases of cigarette smoke exert inhibitory effects on endothelial function via different mechanisms. These may include, or be independent of, elevated cellular free radical levels. Interestingly, acrolein, a chemical constituent of the vapor phase of cigarette smoke, induced free radical production in endothelial cells 45,46 and inhibited endothelial migration in vitro. 47 This could potentially explain the involvement of free radicals in the inhibition of migration by the vapor phase but not in our studies examining the particulate phase.
A further potential explanation for the differential role of elevated free radical levels in the inhibition of endothelial migration is that PM and aqueous smoke extracts could both induce the production of free radicals by different sources or in different intracellular compartments. For example, there are many reports in the literature of a role for the plasmalemmal NADPH oxidase in mediating numerous cellular effects of aqueous cigarette smoke extracts. 48 Furthermore, evidence suggests that cigarette smoke particulate extracts can modulate mitochondrial function 49 and elevate ROS production in cultured endothelial cells. 50 This is not an entirely clear picture as there are also reports, for example, of particulate extracts regulating NADPH oxidase activity. 42 Likely, the source of ROS may have other dependencies such as cell type, length of exposure, and concentration of the extract used. Despite this however, one could hypothesize that both aqueous and tar extracts cause ROS production at different cellular locations and that due to tight spatial control of ROS levels the localization of the particulate-induced ROS elevation makes it unable to regulate migration. The effects of the particulate phase extracts on migration therefore would be via a different and as yet unexplained mechanism but independent of elevated ROS levels. Further studies are required to test this hypothesis and to delineate the different mechanisms underlying the inhibition of endothelial migration by aqueous and particulate phase cigarette smoke extracts. Finally, it is also worthy to note that in the article by Michaud and coworkers, 29 a Boyden chamber assay was used in which endothelial migration was assessed by monitoring chemotactic cell passage through a micropore membrane. Potentially, therefore, the different underlying mechanism and end point (migration rate in a scratch assay) measured in our studies could also give rise to differences in how these assays respond to cigarette smoke extracts.
There is currently little information concerning which specific cigarette smoke toxicants may be involved in elevating CVD risk. When considering the particulate phase toxicants that are present in different amounts in the CC6 and TSS6 cigarettes, both cadmium and benzo(a)pyrene were reduced by similar amounts and which corresponded to the degree of improvement of endothelial migration in cells exposed to TSS6 compared to CC6 PM. Individually, both of these toxicants have been shown previously to have an inhibitory effect on endothelial migration in vitro. 51,52 However, further consideration both of the levels of 3R4F smoke toxicants and of the effects of 3R4F PM on endothelial migration may argue against cadmium and benzo(a)pyrene having a role, since the relative levels of these toxicants in the smoke of the different cigarettes does not fully correlate with the observed migration rates. Our data may also rule out a role for nicotine as a smoke constituent responsible for the differences in migration observed in our studies, since these differences were still observed following normalization of data to nicotine levels. Further, migration rates were similar in cells exposed to CC6 and 3R4F cigarette smoke, which contained markedly different nicotine levels. Further studies in which PM nicotine levels themselves are normalized by spiking PM with nicotine are suggested in order to support this finding. Taking all of this together, while our data do demonstrate that cigarettes with different toxicant yields may exert different biological effects, further experimentation is required before we can draw any conclusions on the specific toxicants responsible for these differences.
In summary, cigarette smoke PM inhibited endothelial migration in an in vitro model of a pathogenic process important in CVD initiation and progression. This effect was modified by exposing cells to smoke from cigarettes with different toxicant yields. While our data suggest a lack of a role for free radical production in the observed effects and as such the mechanisms remain to be elucidated, our data do demonstrate that smoke toxicant reduction can modify a biological response to cigarette smoke and that in vitro disease models possess the ability to detect such changes in biological effects. Further studies are required to fully understand which toxicants are involved in pathogenesis and to explore the clinical implications of toxicant reductions.
Footnotes
Authors' Note
Declaration of Conflicting Interests
The author(s) declared the following potential conflict of interest with respect to the research, authorship, and/or publication of this article: All authors either are currently or were employees of British American Tobacco Group Research and Development. IMF and EB hold stock in British American Tobacco.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: British American Tobacco Group Research and Development.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.