
Abstract
Introduction
The objective of this study was to explore the effect of KLF9 on oxidative stress (OS) and NLRP3-mediated inflammation in preeclampsia (PE).
Methods
Lipopolysaccharide (LPS)+adenosine triphosphate (ATP)-induced HTR-8/SVneo cells were used as an in vitro PE inflammation cell model. shRNA was used to interfere with KLF9 expression (sh-KLF9) to assess the transfection efficiency and the effect of KLF9 on cell proliferation, migration, and invasion. ELISA was performed to detect OS-related factors and inflammatory cytokines. Reactive oxygen species (ROS) levels and pyroptosis were analyzed using DCFH-DA and TUNEL staining. LPS and ATP induced HTR-8/SVneo cells were co-transfected with sh-PRDX6/sh-KLF9 to explore the potential regulatory effect of KLF9 on PRDX6.
Results
LPS+ATP stimulation increased KLF9 expression in the PE cell model. Specifically, reducing KLF9 levels alleviated morphological damage and enhanced proliferation, migration, and invasion in the in vitro PE cell models. Moreover, inhibiting KLF9 expression decreased protein expression of NLRP3, GSDMD-N, cleaved caspase-1, and cleaved-IL-1β, suppressing cell death in LPS+ATP-induced HTR-8/SVneo cells. Analysis of OS indicators revealed that downregulating KLF9 expression restrained intracellular ROS production, decreased MDA expression, and increased SOD and CAT levels. KLF9 regulated the transcription of PRDX6 to attenuate OS and pyroptosis. Knockdown of PRDX6 partially abolished the effect of KLF9 downregulation on OS and pyroptosis of LPS+ATP-induced HTR-8/SVneo cells, as evidenced by the inhibition of cell proliferation, migration, and invasion, as well as the enhanced activity of the NLRP3 inflammasome.
Conclusion
Downregulation of KLF9 enhances trophoblast cell invasion and reduces OS and NLRP3 inflammasome activation-mediated pyroptosis.
Introduction
Preeclampsia (PE) is a prevalent pregnancy complication closely linked to elevated maternal and fetal mortality rates.1,2 Studies have suggested that maternal inflammation and impaired placental function are key factors in the development of complications, including preterm delivery, restricted fetal growth, and PE.3,4 These pregnancy complications have been attributed to notable pathophysiological alterations in the placenta, including abnormalities in the villous tree structure and compromised vascular functionality. 5 Growing evidence suggests that the onset of PE is characterized by an imbalance between the production of reactive oxygen species (ROS) and antioxidant defenses, which leads to oxidative stress. This imbalance activates the inflammatory response, resulting in cellular damage and placental dysfunction. 6 Further research is crucial for identifying therapeutic targets for managing PE progression, particularly focusing on mitigating oxidative stress and inflammation.
Kruppel-like factors (KLFs) are a comprehensive family of transcription factors comprising zinc finger domains that markedly influence PE development by modulating cellular differentiation and function.7,8 Although the role of KLF9 in the pathology of various diseases is well-documented; however, its specific contribution to PE remains unexplored. 9 Notably, KLF9 has been shown to exacerbate inflammation and oxidative stress in mouse models of diabetic cardiomyopathy, 10 whereas the knockdown of KLF9 can mitigate myocardial infarction-induced inflammatory damage. 11 In the current study, we aimed to elucidate the role of KLF9 in mediating oxidative stress and inflammation in PE. In human lens epithelial cells, KLF9 was found to suppress the expression of peroxiredoxin 6 (PRDX6), an antioxidative gene involved in the Nrf2 pathway, which is crucial for maintaining ROS balance. 12 Downregulating PRDX6 expression in the placenta of rats with intrauterine growth restriction could trigger oxidative stress, impairing the proliferation and invasion of trophoblasts. 13 A correlation between the cellular responses to ROS and PRDX6 levels has been previously established. PRDX6 plays a role in reducing oxidative stress and inflammation, thereby mitigating LPS-induced acute kidney injury. 14 Moreover, KLF9 may inhibit antioxidant genes, including PRDX6, leading to an imbalance in antioxidant gene expression and increased ROS production.12,15 Moreover, enhanced PRDX6 expression has been linked to reduced NOD-like receptor thermal protein domain-associated protein 3 (NLRP3) inflammasome activity and decreased release of inflammatory cytokines by macrophages. 16 The improper NLRP3 inflammasome activation has been associated with various reproductive disorders and pregnancy-related conditions, notably PE, as it fosters excessive inflammatory responses by releasing cytokines such as interleukin (IL)-β and IL-18. 17 Herein, we used lipopolysaccharide (LPS) + adenosine triphosphate (ATP; LPS + ATP)-induced HTR-8/SVneo cells to simulate in vitro PE inflammation models. The functional role of KLF9 in oxidative stress and NLRP3 inflammasome activation in trophoblasts was explored to identify the possible regulatory mechanisms of KLF9 in PE.
Materials and methods
Cell culture and treatment
The HTR-8/SVneo cell line was obtained from the Cell Bank of the Type Culture Collection Committee at the Chinese Academy of Sciences (Shanghai, China), cultured in DMEM (Gibco, Grand Island, NY, USA), supplemented with 10% fetal bovine serum (FBS), 1% penicillin, and 1% streptomycin at 37°C under 5% CO2 in a cell culture incubator.
Subsequently, HTR-8/SVneo cells were incubated with 100, 200, or 400 ng/mL LPS (L2880; Sigma) for 1 or 24 h or treated with 200 ng/mL LPS for 0, 20, 40, or 60 min, or 0, 6, 12, or 24 h, followed by treatment with 5 mM ATP for 45 min. Cells treated with the same volume of phosphate-buffered saline (PBS) served as controls.
HTR-8/SVneo cells were assigned to the following groups: sh-NC (disordered shRNA was transfected into cells, followed by treatment with LPS + ATP; GenePharma, Shanghai); sh-KLF9 (KLF9 expression knockdown plasmid was transfected into cells, followed by treatment with LPS + ATP); and sh-PRDX6 (PRDX6 expression knockdown plasmid was transfected into cells and cells were treated with LPS + ATP) groups. Transfected cells were transfected using LipoFiterTM Transfection Reagent (HanBio, Shanghai). After transfection for 24 h, cells were subjected to LPS and ATP induction as well as subsequent functional assays. All cell-based experiments were performed with a minimum of three independent replicates to ensure the reliability and reproducibility of the results.
Reverse transcription-quantitative PCR (RT-qPCR)
Total RNA was extracted using the TRIZOL reagent (Invitrogen, Carlsbad, CA, USA), followed by reverse transcription using a kit from TaKaRa (Tokyo, Japan). Gene expression was assessed using a LightCycler 480 real-time PCR system (Roche, Indianapolis, IN, USA) in accordance with the reaction conditions specified in the SYBR Green Mix PCR kit protocol (Roche Diagnostics, Indianapolis, IN). Quantitative PCR was conducted in triplicate for each sample, with β-actin as the comparative mRNA standard. Data analysis was performed using the 2−ΔΔCt method. The primer sequences used were as follows: KLF9-forward: 5′-CTGGTTGCTGGGACTGTAGC-3,’ KLF9-reverse: 5′-GTTTTCCAGCTCCCAAACAG-3; PRDX6-forward: 5′-GCATCCGTTTCCACGACT -3′; PRDX6-reverse: 5′-TGCACACTGGGGTAAAGTCC-3,’ Β-actin-forward: 5′-CCAAGCAGGAGTACGACGAG-3,’ Β-actin-reverse: 5′-TTTACGATGGCAGCAACGGA-3.’
Western blot analysis
Protein samples were extracted with RIPA lysis buffer (Beyotime, Shanghai, China) and quantified using a BCA assay kit (Beyotime). The samples were combined with loading buffer (Beyotime) and denatured by immersion in boiling water for 3 min. For electrophoresis, samples were run at 80 V for 30 min until bromophenol blue reached the separating gel, followed by application of 120 V for 1–2 h. Protein transfer was carried out at 300 mA for 60 min in an ice bath. Following membrane transfer, the membrane was briefly washed for 1–2 min, blocked for 1 h at ambient temperature or overnight at 4°C, and then incubated for 1 h on a shaker at ambient temperature with the following primary antibodies: KLF9 (1:1000; ab227920; Abcam), PRDX6 (1:1000; ab133348, Abcam), NLRP3 (ab263899, 1:1000, Abcam), GSDMD (1:2000; ab215203, Abcam), cleaved caspase-1 (1:500; Cat#4199, CST), Cleaved-IL-1β (1:500; Cat#83186, CST), and β-actin (1:1000; ab5694, Abcam). Secondary antibodies were added and incubated at ambient temperature for 1 h, followed by three washes for 10 min each. After adding the development solution to the membrane, the detection was performed using a chemiluminescence imaging system (Bio-Rad, Hercules, CA, USA). The bands were analyzed using ImagePro Plus software (Media Cybernetics, Inc., Maryland, USA).
Cell proliferation analysis
Cell proliferation was assessed as an indicator of cell activity using the Cell Counting Kit-8 (CCK8; 521942, Biosharp, Guangzhou, China) assay. The cells were plated in a 96-well plate (4 × 103 cells/well) and incubated for 24 and 48 h. Cell proliferation in each group at each time point was evaluated using five replicate wells. Subsequently, 10 µL of CCK8 solution was added to each well, followed by incubation in a cell culture incubator. The optical density (OD) at 450 nm was quantified using a microplate reader (BioTek Instruments Inc., Winooski, VE, USA).
Transwell assay
Cell invasion abilities were assessed using a transwell assay. The transwell assay was set up using chambers from Corning (NY, USA), featuring an 8 μm pore size, and coated with Matrigel. Initially, a serum-free solution (0.5 mL) was applied to the chambers, which were then conditioned at 37°C and 5% CO2 for 2 h prior to medium removal. Log-phase cells were prepared as a single-cell suspension and seeded into a six-well plate, followed by incubation under the same conditions until the cells reached 70%–90% confluency. Subsequently, the cells were allocated to corresponding groups and cultured for 24 h. The cells were then trypsinized, rinsed with PBS, and resuspended in serum-free medium, adjusting the concentration appropriately. Transwell chambers were prepared accordingly, with 600 μL of a medium infused with 10% FBS in the lower chamber and 100 μL of the cell suspension in the upper chamber. The assembly was then incubated for 24 h. Post-incubation, non-invaded cells were cleared from the upper side of the membrane. Cells that invaded to the underside were fixed with paraformaldehyde for 20 min and stained with Giemsa solution for visualization. Images were captured using a microscope (Olympus, Tokyo, Japan), with five random fields randomly selected for each sample under high magnification. The number of invaded cells was counted using the ImageJ software (National Institutes of Health, Bethesda, MD).
Scratch assay
Logarithmically growing cells were harvested and prepared as a single-cell suspension before uniform distribution on a six-well plate. Following a 24-h incubation period, cells in each group were subjected to specific treatments based on their designated groups, followed by incubation at 37°C with 5% CO2 for an additional 24 h. Using a sterile pipette tip, scratches were created on the surface of the six-well plate, ensuring that the tip was held as vertically as possible to maintain consistent scratch widths among all groups. Subsequently, the culture medium in wells was aspirated, the cells were washed twice with PBS, and serum-free medium was added for further cultivation. The scratch area at 0 h was documented, photographed, and used as the baseline measurement. The plate was maintained at 37°C under 5% CO2 in an incubator for an additional 24 h to observe and capture images of cell migration.
Measurement of ROS
Intracellular levels of ROS were detected using the fluorescent dye 2′,7′-dichlorofluorescin diacetate (DCFH-DA; Merck, Shanghai). Briefly, cells were seeded in a 96-well plate (5000 cells/well) and incubated with 10 μM DCFH-DA, followed by incubation at 37°C for 30 min. The cells were then washed thrice with PBS to remove any residual DCFH-DA. For each well, the fluorescence intensity was measured using a SYNERGY microplate reader at 530 nm.
TUNEL staining
TUNEL staining was performed in accordance with the instructions of the TUNEL kit (C1088; Biotechnology, Wuhan, China). The treated HTR-8/SVneo cells were treated with TUNEL solution for 30 min at 37°C, and nuclei were stained with DAPI. Cells fixed with 4% paraformaldehyde and permeabilized with 0.3% Triton X-100, followed by incubation with 50 µL TUNEL reaction solution. After washing three times with PBS, the cells were stained with DAPI (1 mg/mL) at room temperature for 3 min. Following another round of washing with PBS, the cells were sealed with an anti-fade reagent. Labeled cells were randomly observed under a fluorescence microscope (Apotome3; Zeiss, Oberkochen, Germany). Image analysis was performed using the ImagePro Plus software (Media Cybernetics, Inc., Maryland, USA), and the TUNEL-positive cells were quantified and normalized to the total number of viable cells to determine the TUNEL-positive rate.
Measurement of lactate dehydrogenase (LDH)
LDH activity (U/g) was measured in HTR-8/SVneo cells using an LDH Activity Assay Kit (D799208; Sangon) according to the manufacturer’s instructions. OD at 450 nm was measured using a microplate reader: LDH activity (U/g) = (Measured OD value - Control OD value) / (Standard OD value - Blank OD value) × Standard concentration (2 μmol/L) / protein concentration of the sample (g/mL).
Enzyme-linked immunosorbent assay (ELISA)
HTR-8/SVneo cells were treated with LPS+ATP or PBS for 48 h and then centrifuged at 1500 g for 10 min at 4°C. Cultured supernatant from HTR-8/SVneo cells was collected for experimental requirements and treated with the appropriate induction or transfection procedures (LPS+ATP, PBS) for 48 h. Specific ELISA kits were employed to detect levels of IL-1β (PI305; Beyotime) and IL-18 (PI558; R&D, Beyotime) in the cultured supernatant.
The activities of catalase (CAT) (S0051; Beyotime), malondialdehyde (MDA) (S0131S; Beyotime), and superoxide dismutase (SOD) in the culture supernatant were detected according to the manufacturer’s guidelines.
Statistical analysis
Data analysis was performed using GraphPad Prism 8 (GraphPad Software, Inc., La Jolla, CA, USA). Data values are expressed as mean ± standard error of the mean (SEM). Differences between any two independent groups were analyzed using Student's t-test. For comparisons between three or more groups, one-way ANOVA was employed, followed by Tukey’s test for post-hoc comparisons. A p-value <.05 was deemed statistically significant.
Results
Increased KLF9 expression in the in vitro model of LPS-induced trophoblasts
To quantify the expression of KLF9 in trophoblasts during the inflammatory state of cells, HTR-8/SVneo cells were stimulated with various LPS concentrations at various time points. Western blot analysis demonstrated a dose-dependent increase in KLF9 expression in HTR-8/SVneo cells following LPS treatment for 1 h (short-term) and 24 h (long-term) (Figure 1(a) and (b)). Subsequent examination of alterations in KLF9 protein expression in HTR-8/SVneo cells following LPS exposure (200 ng/mL) revealed a significant time-dependent increase in LPS levels after exposure for 1 and 24 h, respectively (Figure 1(c) and (d)). Collectively, the in vitro PE trophoblast model exhibited elevated KLF9 expression. An increase in the expression of KLF9 in the LPS and ATP-induced inflammatory model of human trophoblasts. (a, b) HTR-8/SVneo cells were treated with specific concentrations of LPS for 1 h and 24 h. Western blot analysis was conducted to measure the protein expression of KLF9. (c, d) HTR-8/SVneo cells were treated with LPS (200 ng/mL) for specified durations. Western blot analysis was performed to assess the protein expression of KLF9. β-Actin was used as an internal control, and all protein band intensities were quantified and normalized for presentation. Data are presented as mean ± standard deviation (SD). The experiment was repeated independently three times. *p < .05; **p < .01; ***p < .001, and one-way analysis of variance was used for multiple group comparison. LPS: lipopolysaccharide; ATP: adenosine triphosphate.
Knockdown of KLF9 promotes trophoblast cell invasion and migration abilities
To elucidate the potential role of KLF9 in trophoblasts, we designed a specific shRNA targeting KLF9 to modulate its expression. Both RT-qPCR and western blot analyses validated the robust interference efficiency (Figure 2(a) and (b)). Following the transfection of sh-KLF9 and its negative control, sh-NC, into HTR-8/SVneo cells, HTR-8/SVneo cells were treated with 200 ng/mL LPS for 24 h, followed by treatment with 5 mM ATP 45 min to simulate trophoblast inflammation in PE. Changes in the cell morphology were observed under an inverted microscope. In the control group, HTR-8/SVneo cells exhibited a typical cobblestone-like appearance. Following treatment with LPS + ATP, the number of cells gradually decreased, the gaps between the cells increased, and the cells began to shrink and become rounded, with some cells detaching. After co-treatment with LPS + ATP and sh-KLF9, cell shrinkage and rounding were reduced, indicating that the specific knockdown of KLF9 significantly alleviated the LPS + ATP-induced morphological damage to HTR-8/SVneo cells (Figure 2(c)). Based on the CCK8 assay results, KLF9 knockdown restored HTR-8/SVneo cell viability (Figure 2(d)). Furthermore, transwell and scratch assays revealed that KLF9 suppression augmented the invasive and migratory abilities of HTR-8/SVneo cells (Figure 2(e) and (f)). Knockdown of KLF9 enhances invasion and migration capabilities in LPS and ATP treated trophoblasts. (a, b) Specific construction of shRNA targeting KLF9 was transfected into HTR-8/SVneo cells. Levels of KLF9 mRNA and protein expression were analyzed by qRT-PCR and western blot. (c) The effect of KLF9 knockdown on the morphology of LPS+ATP induced HTR-8/SVneo cells was observed using an inverted microscope; (d) CCK8 assay was used to analyze changes in cell viability of HTR-8/SVneo cells transfected with sh-KLF9 after treatment with LPS and ATP. (e) Transwell analysis of changes in cell invasion capacity of HTR-8/SVneo cells transfected with sh-KLF9 after treatment with LPS and ATP. (f) Scratch assay analysis of changes in cell migration capability of HTR-8/SVneo cells transfected with sh-KLF9 after treatment with LPS and ATP, with data presented as mean ± standard deviation (SD). The experiment was independently repeated three times. *p < .05; **p < .01; ***p < .001, and one-way analysis of variance was used for comparisons between multiple groups. LPS: lipopolysaccharide; ATP: adenosine triphosphate.
Knockdown of KLF9 suppresses trophoblast cell oxidative stress and pyroptosis
Additionally, we examined oxidative stress and inflammatory alterations in an in vitro PE model following KLF9 knockdown. Initially, we detected the production of SOD, CAT, and MDA in LPS + ATP-treated HTR-8/SVneo cells. Upon treatment with LPS + ATP, MDA levels gradually increased, whereas those of SOD and CAT decreased (Figure 3(a)–(c)). Furthermore, the introduction of sh-KLF9 markedly reduced MDA levels and an elevation in SOD and CAT levels (Figure 3(d)–(f)). Knockdown of KLF9 enhances oxidative stress in trophoblasts. (a–c) HTR-8/SVneo cells were treated with 200 ng/mL LPS and 5 mM ATP for the indicated time and then the oxidative stress related indicators CAT, SOD and MDA were detected using ELISA kit; (d–f) LPS+ATP treated HTR-8/SVneo cells were used to simulate PE in vitro models. HTR-8/SVneo cells were transfected with sh-NC or sh-KLF9 before LPS+ATP treatment, in which the level of CAT, SOD and MDA was detected by ELISA kit. Data are presented as mean ± standard deviation (SD). The experiment was independently repeated three times. *p < .05; **p < .01; ***p < .001, and one-way analysis of variance was used for comparisons among multiple groups. LPS: lipopolysaccharide; ATP: adenosine triphosphate; CAT: catalase; SOD: superoxide dismutase; MDA: malondialdehyde; PE: preeclampsia.
In the presence of multiple stimuli, ROS can induce inflammation through oxidative stress. Therefore, we measured intracellular ROS levels using DCFH-DA, a cell-permeable probe that emits green fluorescence upon ROS oxidation. After LPS+ATP treatment, we detected a significant increase in ROS fluorescence intensity. However, the diminished fluorescence intensity in sh-KLF9-transfected cells indicated the potential of KLF9 knockdown to suppress ROS production (Figure 4(a) and (b)). KL9F knockdown can suppress the LPS+ATP induced NLRP3 inflammasome activation and pyroptosis in HTR-8/SVneo cells. (a, b) HTR-8/SVneo cells were transfected with sh-NC or sh-KLF9 before LPS+ATP treatment, in which the intracellular ROS levels were measured using the DCFH-DA fluorescent probe. (c, d) Western blot detected the protein level of NLRP3, GSDMD-N, cleaved-Caspase 1 and cleaved-IL-1β in HTR-8/SVneo cells and their corresponding quantitative statistical analysis. (e, f) Cell pyroptosis number detected by TUNEL staining; (g) LDH release; (h) ELISA assay was used to measure the production of IL-18 and IL-1β in specified cell culture medium. Data are presented as mean ± standard deviation (SD). The experiment was independently repeated three times. *p < .05; **p < .01; ***p < .001, and one-way analysis of variance was used for comparisons among multiple groups. LPS: lipopolysaccharide; ATP: adenosine triphosphate; ROS: reactive oxygen species; DCFH-DA: dye 2′,7′-Dichlorofluorescin diacetate; NLRP3: NOD-like receptor family pyrin domain containing 3; GSDMD-N: gasdermin D N-terminal domain; IL-1β: interleukin-1 beta; IL-18: interleukin-18; LDH: lactate dehydrogenase.
To validate the role of KLF9 in NLRP3 inflammasome activation in PE models, we detected NLRP3 inflammasome activation and cell pyroptosis in HTR-8/SVneo cells. The results revealed that stimulation of HTR-8/SVneo cells with LPS + ATP induced NLRP3 inflammasome activation, whereas downregulation of KLF9 attenuated the upregulation of NLRP3, GSDMD-N, cleaved caspase-1 and cleaved-IL-1β (Figure 4(c) and (d)). Moreover, TUNEL staining demonstrated that treatment with LPS + ATP increased TUNEL-positive cell numbers; however, this increase was suppressed in response to transfection with sh-KLF9 (Figure 4(e) and (f)). Considering pyroptosis detection, the LDH assay showed that, compared with cells without LPS and ATP treatment, LPS+ATP-treated HTR-8/SVneo cells had an increased LDH content (Figure 4(g))). According to the ELISA results, KLF9 knockdown also decreased the production of IL-1β and IL-18 (Figure 4(h))). These results indicated that KLF9 suppression could inhibit the NLRP3 inflammasome activation induced by oxidative stress in trophoblasts.
KLF9 negatively regulates PRDX6
Increasing LPS concentrations and treatment durations led to decreased PRDX6 mRNA and protein expression in LPS-induced HTR-8/SVneo cells (Figure 5(a)–(d)); knockdown of KLF9 in LPS-treated HTR-8/SVneo cells led to increased expression of PRDX6 (Figure 5(e) and (f)). These results suggested that KLF9 could negatively regulate PRDX6 transcription. KLF9 binds to the PRDX6 promoter region. (a, b) HTR-8/SVneo cells were treated with the indicated concentrations of LPS for 24 h. PRDX6 mRNA and protein expression level were analyzed by qRT-PCR and western blot. (c, d) HTR-8/SVneo cells were treated with LPS (200 ng/mL) for the indicated times. PRDX6 mRNA and protein expression level were analyzed by qRT-PCR and western blot. (e, f) HTR-8/SVneo cells were transfected with sh-NC or sh-KLF9 plasmids. After cell transfection for 24 h, HTR-8/SVneo cells were exposed to different concentrations (0 or 200 ng/mL) of LPS, and the mRNA and protein expression levels of PRDX6 were analyzed by qRT-PCR and western blot. Data are presented as mean ± standard deviation (SD). The experiment was independently repeated three times. *p < .05; **p < .01; ***p < .001, and one-way analysis of variance was used for comparisons among multiple groups. LPS: lipopolysaccharide.
PRDX6 reverses the effect of KLF9 on trophoblast cell invasion, oxidative stress, and pyroptosis
We further investigated whether the effect of KLF9 on PE was mediated by targeting PRDX6. HTR-8/SVneo cells were co-transfected with sh-KLF9 and sh-PRDX6 prior to LPS + ATP treatment. Additionally, cells transfected with sh-KLF9 showed significantly elevated PRDX6 expression, but this elevation was partially reversed in response to further co-transfection with the sh-PRDX6 plasmid (Figure 6(a) and (b)). Functional assays of cell biological functions demonstrated that the suppression of PRDX6 expression negated the enhancing effect of sh-KLF9 on trophoblast cell invasion and migration (Figure 6(c) and (d)). PRDX6 reverses the impact of KLF9 on migration and invasion of trophoblasts. HTR-8/SVneo cells were co-transfected with sh-KLF9 and sh-PRDX6 followed by treatment with LPS+ATP. (a, b) qRT-PCR and Western blot were performed to analyze the expression of PRDX6 mRNA and protein in HTR-8/SVneo cells; (c) Transwell assay was employed to assess cell invasion capability; (d) Scratch assay was employed to evaluate cell migration ability; The data are presented as the mean ± standard deviation (SD). All experiments were independently repeated three times. *p < .05; **p < .01; ***p < .001, one-way analysis of variance was used for comparisons among multiple groups. LPS: lipopolysaccharide; ATP: adenosine triphosphate.
Assessment of intracellular ROS, MDA, SOD, and CAT levels indicated that knockdown of PRDX6 abrogated the inhibitory effect of sh-KLF9 on trophoblast oxidative stress levels (Figure 7(a)–(d)). Additionally, western blot analysis showed that the knockdown of PRDX6 notably elevated NLRP3 inflammasome expression and enhanced levels of GSDMD, cleaved caspase-1 and cleaved-IL-1β. Detection of cell death, LDH release, and IL-1β and IL-18 levels confirmed that knockdown of PRDX6 promoted cell death, increased LDH release, and restored IL-1β and IL-18 levels (Figure 7(e)-(h)). These findings suggested that PRDX6 abolishes the effects of KLF9 on cell invasion, oxidative stress, and pyroptosis in trophoblasts. PRDX6 reverses the impact of KLF9 on trophoblast cell oxidative stress and inflammation. (a) The intracellular ROS levels were measured using the DCFH-DA fluorescent probe; (b–d) Determination of intracellular MDA, SOD, and CAT level; (e) Western blot analysis of the protein expression level of NLRP3, GSDMD-N, cleaved caspase-1 and cleaved caspase-1L-β in HTR-8/SVneo cells and their corresponding quantitative statistical analysis; (f) Cell pyroptosis number detected by TUNEL staining; (g) LDH release; (h) ELISA assay was used to measure the production of IL-18 and IL-1β in specified cell culture medium. The data are presented as the mean ± standard deviation (SD). All experiments were independently repeated three times. *p < .05; **p < .01; ***p < .001, one-way analysis of variance was used for comparisons among multiple groups. CAT: catalase; SOD: superoxide dismutase; MDA: malondialdehyde; ROS: reactive oxygen species; LDH: lactate dehydrogenase; DCFH-DA: dye 2′,7′-dichlorofluorescin diacetate; NLRP3: NOD-like receptor family pyrin domain containing 3; GSDMD-N: gasdermin D N-terminal domain; IL-1β: interleukin-1 beta; IL-18: interleukin-18.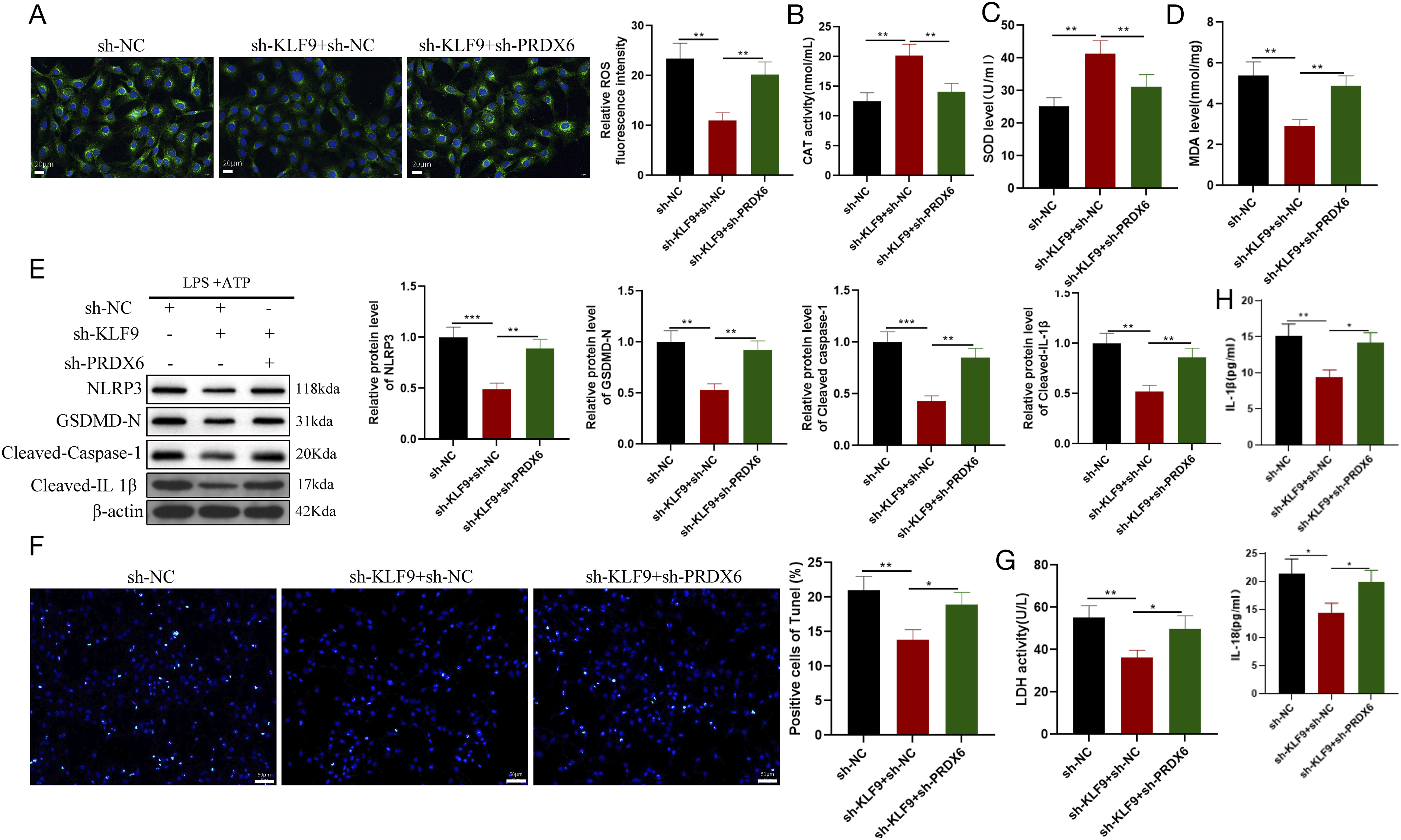
Discussion
In the current study, we identified the transcription factor KLF9 as a novel regulator of cellular oxidative stress and NLRP3 inflammasome-mediated pyroptosis in PE. Our data suggests that KLF9 was upregulated in LPS-stimulated trophoblasts. KLF9 knockdown promoted trophoblast cell proliferation, migration, and invasion and played a critical role against oxidative stress and NLRP3 inflammasome activation-mediated pyroptosis. However, the protective effect of KLF9 knockdown was negated by PRDX6 knockout in trophoblasts (Supplemental Figure 1).
As a transcription factor, KLF9 regulates several important cellular biological processes, such as differentiation, proliferation, development, survival, and responses to external stress.10,18 Herein, our findings revealed an increase in KLF9 expression in LPS+ ATP-treated HTR8/SVneo cells, whereas KLF9 knockdown potentiated the invasion and migration of HTR-8/SVneo cells, in addition to suppressing oxidative stress and inhibiting pyroptosis. These results align with those of a recent study showing that KLF9 activation leads to increased CYP1A1 expression, which is linked to poor pregnancy outcomes and compromised functions in HTR8/SVneo cells, such as proliferation and oxidative stress management. 19 Our data confirmed that KLF9 silencing in LPS+ATP-induced trophoblasts reduced NLRP3 inflammasome activation, as evidenced by the decreased expression of NLRP3, GSDMD-N, cleaved caspase-1, and cleaved-IL-1β. This reduction is notable, given that GSDMD and cleaved caspase-1 are key indicators of deactivation of NLRP3 inflammasome, a complex involved in pro-inflammatory cytokine production.20–22 NLRP3 inflammasome activation in trophoblasts is associated with the pathogenesis of placental inflammation, and elevated levels of ROS can lead to the NLRP3 inflammasome activation.23,24 KLF9 is known to elevate ROS levels in various cultured cells and mouse models by binding to and altering the expression of ROS metabolism-related genes. 25 Upon formation, the NLRP3 inflammasome triggers caspase-1 activation, leading to gasdermin D-dependent pyroptosis, which, in turn, facilitates the release of IL-1β and IL-18 26 and initiates pyroptosis. 27 In our study, the detection of the LDH content, inflammatory IL-1β and IL-18 levels, and TUNEL-stained positive cells highlighted that KLF9 knockdown can suppress NLRP3 inflammasome activation-induced pyroptosis in PE cell models. Consistent with the results of this study, the reduction of KLF9 expression in the inflammatory response has been previously reported in macrophages following myocardial infarction 11 and in LPS-induced RAW264.7 cells. 28 Collectively, these findings suggest that KLF9 activates the NLRP3 inflammasome to enhance oxidative stress and pyroptosis in LPS + ATP-treated trophoblasts, highlighting its potential as a therapeutic target for PE.
To clarify the mechanism underlying this phenomenon, we analyzed factors downstream of KLF9 and determined whether KLF9 can negatively regulate the expression of PRDX6 in HTR-8/SVne cells. Our experimental findings further demonstrated that reducing PRDX6 levels could mitigate the effects of KLF9 knockdown on trophoblast cell function. Of note, PRDX6 expression was upregulated in the absence of KLF9. Furthermore, PRDX6 knockdown was found to eliminate the protective effect of KLF9 deficiency on SH-SY5Y cell survival and reverse the reduction in ROS production in cells and mitochondria. 29 Compared with control cells, PRDX6 siRNA-treated rabbit oviduct epithelial cells showed increased expression of oxidative stress- and inflammation-related proteins. 30 Additionally, PRDX6 deficiency reportedly leads to NLRP3 inflammasome activation and subsequent pyroptosis in aging or redox-active cells, a process that can be reversed by PRDX6 overexpression. Accordingly, oxidative stress can increase KLF9 expression in PRDX6-deficient mouse lens epithelial cells, thereby promoting NLRP3 transcription in a KLF9-dependent manner. Conversely, reducing ROS levels through PRDX6 overexpression or KLF9 knockdown diminishes inflammatory responses.31,32 Collectively, these findings highlight the crucial role of PRDX6 in controlling trophoblast cell function, oxidative stress, and pyroptosis, thus establishing PRDX6 deficiency as a contributing factor to KLF9-induced oxidative stress and pyroptosis in PE.
In this study, a previously unidentified regulatory pathway, KLF9/PRDX6/NLRP3, was identified as a crucial factor for regulating trophoblast cell invasion, oxidative stress, and pyroptosis in PE. The findings of this study offer new perspectives that could advance the understanding of the functional significance of KLF9 in PE, as well as potential biomarkers for therapeutic advancements in PE management.
Supplemental material
Supplemental Material - KLF9 mediates NLRP3 inflammasome and reactive oxygen species to mediate pyroptosis in trophoblasts
Supplemental Material for KLF9 mediates NLRP3 inflammasome and reactive oxygen species to mediate pyroptosis in trophoblasts by Qian Li and Min Chen in Journal of Human & Experimental Toxicology.
Footnotes
Author contributions
Min Chen conceived the ideas. Min Chen and Qian Li designed the experiments, performed the experiments, analyzed the data, provided critical materials, and wrote the manuscript. Both authors have read and approved the final version for publication.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Supplemental Material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.