
Abstract
Objectives
Methamphetamine (MA) abuse is a serious social problem worldwide. Cardiovascular complications were the second leading cause of death among MA abusers. We aimed to clarify the effects of MA on myocardial injury, oxidative stress, and apoptosis in myocardial cells and to explore the potential mechanism of nuclear factor-erythroid factor 2-related factor 2 (Nrf2) in MA-induced oxidative stress and apoptosis.
Methods
An acute cardiac toxicity model of MA was established by intraperitoneal injection of MA (2 mg/kg) for 5 days. Nrf2 activation (by sulforaphane (SFN) 1 h before MA injection) and Nrf2 gene knockout were performed to explore the regulatory effects of Nrf2 on cardiac toxicity.
Results
The protein expressions of Nrf2 (p < .001) and heme oxygenase-1 (HO-1) were increased (p < .01), suggesting that MA activated the Nrf2/HO-1 pathway. In the MA group, cardiac injury score (p < .001) and cardiac troponin I (cTnI) protein expression increased (p < .01). Malondialdehyde (MDA) content increased (p < .001), superoxide dismutase (SOD) activity decreased (p < .05). Protein expressions of Caspase-3 (p < .001) and Bax (p < .001) increased, and Bcl-2 decreased (p < .001) as well. These changes were reversed by activation of Nrf2 but became more pronounced after Nrf2 knockout, suggested that the activation and knockout of Nrf2 attenuated and aggravated MA-induced myocardial injury, oxidative stress and apoptosis in myocardial cells, respectively.
Conclusions
MA administration induced myocardial injury, oxidative stress, and apoptosis in mice. Nrf2 attenuated MA-induced myocardial injury by regulating oxidative stress and apoptosis, thus playing a protective role.
Introduction
Methamphetamine (MA) abuse is a huge social problem worldwide. 1 MA is a cationic molecule and chiral compound with phenylethylamine as its core 2 and is a widely abused illegal psychostimulant with highly addictive and physical toxicity. 3 As a sympathomimetic amine, MA exerts a wide range of adverse effects on multiple organ systems. 4 Cardiovascular complications are the second leading cause of death among MA abusers 5 and include hypertension, aortic coarctation, acute coronary syndrome, pulmonary hypertension, and methamphetamine-related cardiomyopathy.6–8 Possible mechanisms by which MA-induced cardiotoxicity causes cardiovascular complications include oxidative stress and apoptosis. 9 MA can trigger cardiac toxicity and cardiovascular disease by inducing reactive oxygen species (ROS) production, redox imbalance, 10 and oxidative stress imbalance. 11 MA can also induce Rho-associated protein kinase 2 (ROCK2), which plays an important role in regulating apoptosis of myocardial cells.12–14 To date, however, the mechanism by which MA induces oxidative stress and apoptosis is not clear in the field of drug injury, and further research is needed.
Nuclear factor-erythroid factor 2-related factor 2 (Nrf2) is a key transcription factor that regulates the functional expression of oxidative stress-related genes and endogenous antioxidants. 15 Under oxidative damage, Nrf2 activates the transcription sequence of cytoprotective genes through downstream elements. 16 Nrf2 is also important in protecting cardiac mitochondria and maintaining or renewing energy metabolism after tissue damage. 17 Heme oxygenase-1 (HO-1) is an inducible cytoprotective enzyme that plays an important role in the regulation of oxidative stress and apoptosis. 18 HO-1 activity is closely associated with the development and progression of a variety of diseases, especially cardiovascular diseases. 19 Nrf2 can directly regulate HO-1 promoter activity and exert antioxidant effects, 20 with both playing protective roles in cardiovascular diseases. 21 Nrf2 can also affect apoptosis and ROCK2 expression.22,23 Therefore, the Nrf2/HO-1 pathway may be an important way to protect cardiomyocytes from oxidative stress and apoptosis. However, whether the Nrf2/HO-1 pathway plays a key role in MA-induced oxidative stress and apoptosis remains to be determined.
Therefore, we hypothesized that the Nrf2/HO-1 pathway might attenuate MA-induced myocardial injury by regulating oxidative stress and apoptosis. To verify this hypothesis, we established a mouse model of MA-induced cardiotoxicity and assessed MA-induced toxic damage to the heart. Using the Nrf2-specific agonist sulforaphane (SFN) to activate Nrf2 and gene knockout technology to knock out the Nrf2 gene, we further investigated the regulatory role of Nrf2 in MA-induced oxidative stress and apoptosis. Our results should provide a reference for further studies on MA-induced toxicity damage and provide support for in-depth exploration of the potential molecular mechanisms underlying MA-induced cardiac toxicity.
Material and methods
Animals
The study was conducted in accordance with the Basic & Clinical Pharmacology & Toxicology policy for experimental and clinical studies. 24 Adult male C57BL/6J mice (18–20 g) were purchased from the Hunan Sleek Jingda Laboratory Animal Company. Specific-pathogen-free (SPF) Nrf2-/- knockout (Nrf2-KO) mice were purchased from the Taicang Saye Model Biology Research Center Company. Knockout mice were raised at the Department of Experimental Animal Science, Kunming Medical University. All mice were housed in plastic cages (six mice per cage) with free access to pure water and food. Mice were placed in animal rooms for 3 days under a controlled ambient temperature (18°C–26°C) with alternating cycles of 12 h light and 12 h dark. For formal experiments, the mice were grouped and housed in a single cage. All animal care and experimental procedures were approved by the Laboratory Animal Ethics Committee of Kunming Medical University (approval code: kmmu2021596).
Establishment of animal models
The MA (purity >98%) was legally provided by the Institute of Criminal Science and Technology of Yunnan Provincial Public Security Department. The SFN (MUST-19121910) was purchased from Chengdu Manster Biotechnology Company. The mice were randomly divided into six equal-sized groups (n = 6): control (10 mL/kg saline, ip), MA (2 mg/kg, ip), SFN (10 mg/kg, ip), SFN (10 mg/kg) + MA (2 mg/kg, ip), Nrf2-KO (10 mL/kg, saline), and Nrf2-KO + MA (2 mg/kg) groups. The doses of MA and SFN were based on previous research.25,26 The mice received SFN once a day or MA twice a day for 5 days. The SFN was administered pre-emptively 1 h before the MA injection for pre-intervention. The animal model scheme is shown in Figure 1. After anesthetize mice with ether in a closed glass container, the mice were euthanized by spinal dislocation method. Cardiac tissue and blood samples were collected, then either fixed in paraformaldehyde or stored at −80°for subsequent research. Experimental protocol of mouse model.
Serological tests (n = 6)
Serum samples were isolated from the control and MA group of mice to measure Myocardial enzymes, included lactate dehydrogenase (LDH) and creatine kinase isoenzymes (CK-MB). In brief, blood was first collected using the femoral vein blood collection method, then stood in a water bath at 37°C (>1 h) and centrifuged (4000 rpm, 15 min) to obtain the supernatant. The serum was sent to the Department of Zoology at Kunming Medical University. LDH and CK-MB were detected using LDH (2030-717, Ryan, Ningbo, China) and CK-MB (2020-717, Ryan, Ningbo, China) assay kits according to the experimental instructions. The expression levels of both were detected using an automatic biochemistry analyzer (AU480, Beckman Coulter, USA).
Hematoxylin-eosin (H&E) staining (n = 6)
The collected mouse hearts were first fixed in paraformaldehyde, then dehydrated and transparentized in a dehydrator, embedded in wax, sliced to a thickness of 5 μm, and stained with H&E. A digital pathological section scanning system (KF-PRO-005-EX, KFBIO, Ningbo, China) was used to examine the cardiac tissue sections to assess myocardial injury. Histological scoring was conducted by two expert pathologists in a blinded manner. 27 Score based on the percentage of damage (myocardial fiber edema, degeneration, necrosis, and inflammatory cell infiltration) involved in each visual field to the total visual field: 0 (none), means no myocardial lesion; 1 means lesions involving <25%; 2 means lesions involving 25 to 50%; 3 means lesions involving 50 to 75%; 4 means lesions involving >75%. 28
Malondialdehyde (MDA) and superoxide dismutase (SOD) assays (n = 6)
Cardiac tissue (0.02 g) was added with nine volumes of saline according to the weight (g):volume (ml) ratio of 1:9. The MDA content and SOD activity, as oxidative stress-related indices, were measured using MDA (TBA method, A003-1-2, Nanjing Jiancheng Institute of Biological Engineering, China) and SOD assay kits (WST-1 method, A001-3, Nanjing Jiancheng Institute of Biological Engineering, China), respectively, according to the provided instructions.
Western blot analysis (n = 6)
Cardiac tissue (left ventricle) was extracted from all mice in each group, and proteins were extracted after splitting the tissue with lysate (Beyotime, P0013B, Shanghai, China) containing protease and phosphatase inhibitors. The samples were separated with equal amounts of protein (25 μg) using 10% polyacrylamide gels. Protein bands were transferred to polyvinylidene difluoride (PVDF) membranes, which were then blocked with 5% skimmed milk for 2 h at room temperature. The membranes were incubated overnight at 4°C with primary antibodies, including rabbit anti-Nrf2 (1:1 000, 16396-1-AP, Proteintech, USA), rabbit anti-HO-1 (1:1 000, 10701-1-AP, Proteintech, USA), rabbit anti-Caspase-3 (1:1 000, 9662S, Cell Signaling Technology, USA), rabbit anti-cTnI (1:1 000, 21652-1-AP, Proteintech, USA), rabbit anti-Bcl-2 (1:5 000, ab196495, Abcam, UK), rabbit anti-ROCK2 (1:3 000, 21645-1-AP, Proteintech, USA), rabbit anti-Bax (1:6 000, 50599-2-Ig, Proteintech, USA), and rabbit anti-GAPDH (1:1 000, BL006B, Biosharp, China), then incubated with horseradish peroxidase (HRP)-coupled anti-rabbit secondary antibodies (1:5 000, 7074S, USA, Cell Signaling Technology, USA) for 2 h at room temperature. The membranes were examined using an enhanced chemiluminescence plus detection kit (Biosharp, BL520B, Shanghai, China). Protein bands were visualized using the Bio-Rad imaging system (GelDoc XR+, Bio-Rad, USA) and detected using ImageJ software. GAPDH was used as an internal reference.
Statistical analysis
Data were processed using SPSS v21.0 (IBM SPSS, Chicago, USA). All data were expressed as mean ± standard deviation (SD). The graphs were constructed using GraphPad Prism v6.0 (GraphPad Software, USA). For analysis of experimental data, independent sample t-test was used to compare two groups and one-way analysis of variance (ANOVA) was used to compare multiple groups, followed by Tukey’s post hoc test. A p-value of less than 0.05 was considered statistically significant.
Results
MA-induced myocardial injury, oxidative stress, and apoptosis, with involvement of Nrf2
MA-induced myocardial injury
The MA group showed various pathological changes, including disorganized myocardial fiber arrangement, deep cytoplasmic staining, and cell aggregation (Figure 2(a)). Compared with the control group, cardiac injury score (***p < .001) and weight (*p < .05) was significantly increased in the MA group (Figure 2(b) and (c)). In addition, the concentrations of CK-MB (*p < .05) and LDH (*p < .05) were significantly elevated in the MA group (Figure 2(d)), as was cardiac expression of the cTnI protein (**p < .01) (Figure 2(e)). These results suggest that MA can induce myocardial injury in mice. MA-induced myocardial injury, oxidative stress, and apoptosis. (a): Cardiac injury was assessed by H&E staining. Bar = 50 µm, magnification 400×. Histopathological changes in myocardium after MA administration, including disorganized myocardial fiber arrangement (red arrow), and cell aggregation (black arrow). (b): Semi-quantitative histopathological score. (c): Comparison of cardiac weights between control and MA group mice. (d): Comparison of serum levels of LDH and CK-MB in mice after MA administration. (e) and (f): Expression levels of cTnI, Caspase-3, Bax, Bcl-2, Nrf2, and HO-1 proteins in cardiac tissue of mice detected by western blotting after MA administration. Data are expressed as mean ± SD. *p < .05, **p < .01, ***p < .001 vs Control.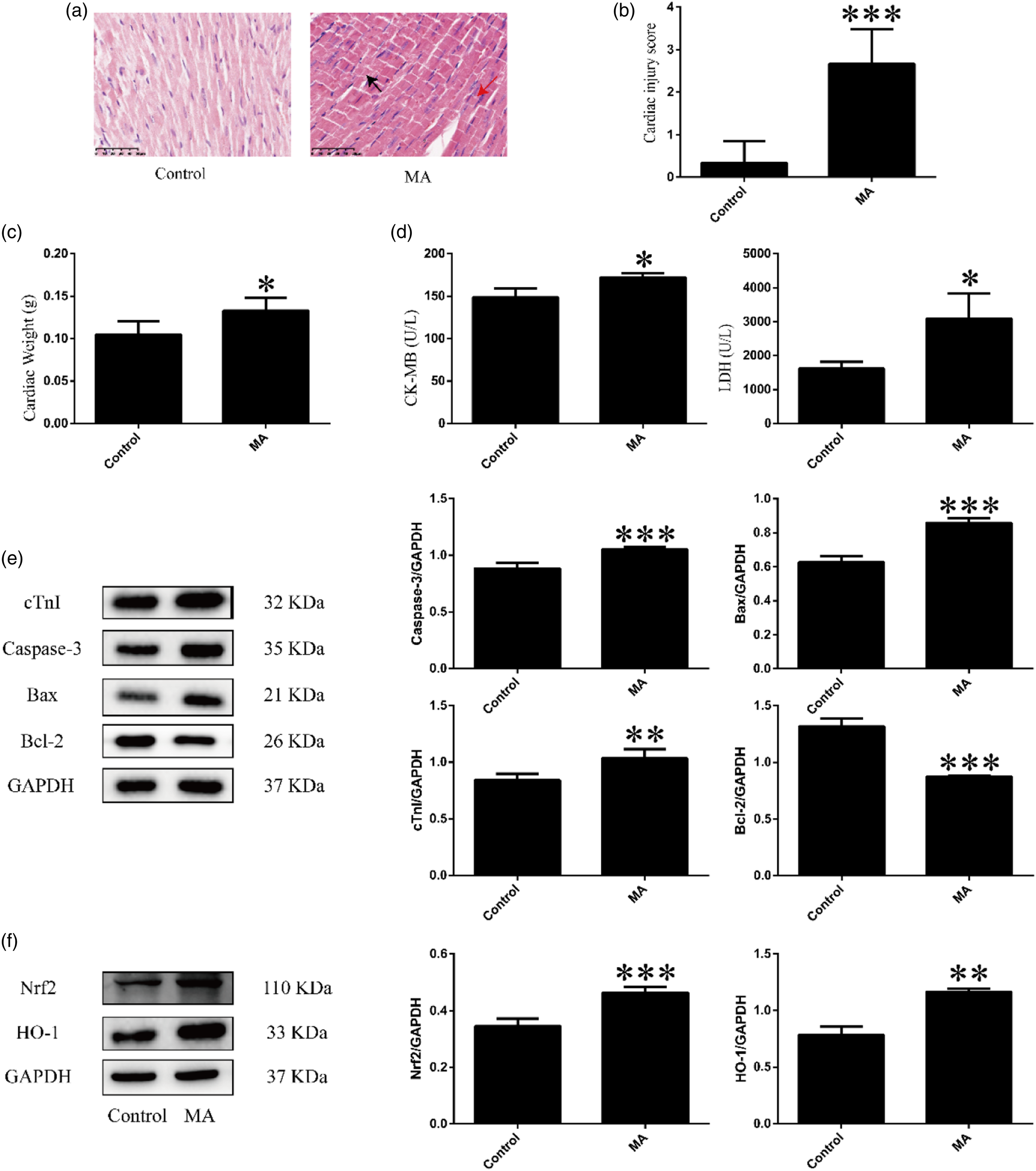
MA-induced oxidative stress and apoptosis of myocardial cells
Compared with the control group, MDA content was significantly increased (***p < .001) and SOD activity was significantly decreased (*p < .05) in the MA group (Figure 3(a) and (c)), suggesting that MA induced oxidative stress in the myocardial cells. Effects of Nrf2 activation or knockout on MA-induced oxidative stress in mice. (a): Changes in MDA content in cardiac tissue of mice after MA administration. (b): Changes in MDA content in cardiac tissue of mice after activation or knockout of Nrf2. (c): Changes in SOD activity in cardiac tissue of mice after MA administration. (d): Changes in SOD activity in cardiac tissue of mice after activation or knockout of Nrf2. Data are expressed as mean ± SD. *P < 0.05,**P < 0.01,***P < 0.001 vs Control; #P < 0.05, ##P < 0.01, ###P < 0.001 vs MA; &&P < 0.01, &&&P < 0.001 vs SFN; $$P < 0.01 vs Nrf2-KO.
Compared with the control group, the protein expression levels of Caspase-3 (***p < .001) and Bax (***p < .001) were increased, while expression of the anti-apoptotic factor Bcl-2 was decreased (***p < .001) in the MA group (Figure 2(e)), suggesting that MA can induce apoptosis of myocardial cells.
MA activation of the Nrf2/HO-1 pathway
Compared with the control group, the protein expression levels of Nrf2 (***p < .001) and its downstream factor HO-1 (**p < .01) were significantly increased in the MA group (Figure 2(f)), suggesting that MA can activate the Nrf2/HO-1 pathway.
Attenuation of MA-induced myocardial injury, oxidative stress, and apoptosis after Nrf2 activation
Attenuation of MA-induced myocardial injury after Nrf2 activation
To verify Nrf2/HO-1 pathway activation after SFN pre-treatment, we detected the expression levels of Nrf2 and HO-1 proteins in mouse hearts by western blotting (Figure 4). Compared with the control group, the protein expression levels of Nrf2 (***p < .001) and HO-1 (***p < .001) were significantly increased in the SFN group. Compared with the MA group, the protein expression levels of Nrf2 (##p < .01) and HO-1 (###p < .001) were significantly increased in the SFN + MA group. These results suggest that the Nrf2/HO-1 pathway may be involved in MA-induced myocardial injury. Expression levels of Nrf2 and HO-1 proteins. Expression levels of Nrf2 and HO-1 proteins in cardiac tissue of mice detected by western blotting after SFN and/or MA administration. Data are expressed as mean ± SD. *p < .05, **p < .01, ***p < .001versus Control; ##p < .01, ###p < .001 vs MA.
After Nrf2 activation, the MA-induced myocardial histopathological changes were alleviated, and the protein expression level of cTnI was reduced.
Myocardial histopathological changes were observed in the MA group, including nucleolysis, nuclear fission, myocardial fiber misalignment, and mild myocardial fibrosis. These myocardial changes were significantly attenuated in the SFN + MA group compared with the MA group. Meanwhile, the cardiac injury score also showed a similar trend of change (Figure 5(a) and (b)). Compared with the control group, the protein expression level of cTnI was increased in the MA group (*p < .05). Compared with the MA group, the protein expression level of cTnI was significantly decreased in the SFN + MA group (###p < .001) (Figure 5(c)). These results suggest that Nrf2 plays a protective role in MA-induced myocardial injury. Effects of Nrf2 activation on MA-induced myocardial injury. (a): Cardiac injury was assessed by H&E staining. Bar = 50 µm, magnification 400×. Histopathological changes in myocardium after SFN and/or MA administration, including nuclear lysis, nuclear fission (black arrow), and myocardial fiber misalignment (red arrow) in MA group, and mild myocardial fiber disorder in SFN + MA group. (b): Semi-quantitative histopathological score. (c): Protein expression level of cTnI detected by western blotting after SFN and/or MA administration. Data are expressed as mean ± SD. *P < 0.05 vs Control; ###P < 0.001 vs MA.
Attenuation of MA-induced oxidative stress after Nrf2 activation
We detected the content of oxidative stress marker MDA and activity of anti-oxidative stress marker SOD, as shown in Figure 3(b) and (d). Compared with the control group, MDA content was significantly increased (***p < .001) and SOD activity was significantly decreased (*p < .05) in the MA group. Compared with the MA group, MDA content was significantly decreased (##p < .01) and SOD activity was significantly increased (###p < .001) in the SFN + MA group. These results suggest that activation of Nrf2 can inhibit MA-induced oxidative stress.
Attenuation of MA-induced apoptosis after Nrf2 activation
As shown in Figure 6, the protein expression levels of Caspase-3 (*p < .05) and Bax (*p < .05) were increased, while the protein expression level of Bcl-2 was significantly decreased (**p < .01) in the MA group compared with the control group. Compared with the MA group, the protein expression levels of Caspase-3 (###p < .001) and Bax (##p < .01) were decreased, while the protein expression level of Bcl-2 was increased (##p < .01) in the SFN + MA group. We also detected the expression levels of ROCK2, a factor closely related to apoptosis. Results showed that ROCK2 protein expression was increased (*p < .05) in the MA group compared with the control group, but decreased (##p < .01) in the SFN + MA group compared with the MA group, indicating that Nrf2 activation can inhibit MA-induced elevation of ROCK2. Thus, these results suggest that Nrf2 activation can inhibit MA-induced apoptosis. Effects of Nrf2 activation on MA-induced apoptosis. Protein expression levels of Caspase-3, Bax, Bcl-2, and ROCK2 in cardiac tissue of mice after SFN and/or MA administration. Data are expressed as mean ± SD. *p < .05, **p < .01 vs Control; ##p < .01, ###p < .001 vs MA.
Aggravation of MA-induced myocardial injury, oxidative stress, and apoptosis after Nrf2 knockout
Aggravation of MA-induced myocardial injury after Nrf2 knockout
MA induced various histopathological changes in the heart, including nuclear aggregation in patches and myocardial hypertrophy. Compared with the MA group, myocardial histopathological changes not only increased in the Nrf2-KO + MA group, but myocardial fiber disorder and misalignment were also observed (Figure 7(a)). Meanwhile, the cardiac injury score (#p < .05) showed an aggravation of MA-induced myocardial injury after Nrf2 knockout (Figure 7(b)). Compared with the control group, the protein expression level of cTnI was increased (*p < .05) in the MA and Nrf2-KO groups. Compared with the MA group, the protein expression level of cTnI was increased (##p < .01) in the Nrf2-KO + MA group (Figure 7(c)). These results suggest that knockout of Nrf2 can exacerbate MA-induced myocardial injury. Effects of Nrf2 knockout on MA-induced myocardial injury. (a): Cardiac injury was assessed by H&E staining. Bar = 50 µm, magnification 400×. Histopathological changes in myocardium after Nrf2 knockout and/or MA administration, including nuclear aggregation into sheets (black arrow) and myocardial hypertrophy in MA group, and disorganized and misaligned myocardial fibers (red arrow) in Nrf2-KO + MA group. (b): Semi-quantitative histopathological score. (c): Expression level of cTnI protein after Nrf2 knockout and/or MA administration. Data are expressed as mean ± SD. *P < 0.05 vs Control; ##P < 0.01 vs MA; $$$P < 0.001 vs Nrf2-KO.
Aggravation of MA-induced oxidative stress after Nrf2 knockout
Compared with the MA group, MDA content was significantly increased (###p < .001) and SOD activity was significantly decreased (#p < .05) in the Nrf2-KO + MA group (Figure 3(b) and (d)), suggesting that knockout of Nrf2 can exacerbate MA-induced oxidative stress.
Aggravation of MA-induced apoptosis after Nrf2 knockout
As shown in Figure 8, the protein expression levels of Caspase-3 (**p < .01) and Bax (*p < .05) were significantly increased, while the protein expression level of Bcl-2 was significantly decreased (***p < .001) in the MA and Nrf2-KO groups compared with the control group. Compared with the MA group, the protein expression levels of Caspase-3 (#p < .05) and Bax (##p < .001) were increased, while the protein expression level of Bcl-2 was decreased (###p < .001) in the Nrf2-KO + MA group. Furthermore, compared with the control group, the protein expression level of ROCK2 was increased (**p < .01) in the MA group. Compared with the MA group, the protein expression level of ROCK2 was increased (###p < .001) in the Nrf2-KO + MA group, suggesting that Nrf2 knockout increases MA-induced elevation of ROCK2. Thus, the above results suggest that Nrf2 knockout can exacerbate MA-induced apoptosis. Effects of Nrf2 knockout on MA-induced apoptosis. Protein expression levels of Caspase-3, Bax, Bcl-2, and ROCK2 in cardiac tissue of mice detected by western blotting after SFN and/or MA administration. Data are expressed as mean ± SD. *p < .05, **p < .01, ***p < .001 vs Control; #p < .05, ###p < .001 vs MA; $$p < .01, $$$p < .001 vs Nrf2-KO.
Discussion
Myocardial injury caused by MA can be reflected by changes in cardiac weight, 29 myocardial pathology 30 and histopathological score, myocardial enzymes, 31 and cTnI protein expression. 32 In the present study, we found that cardiac weight was significantly higher in the MA group. Furthermore, various histopathological changes, such as myocardial hypertrophy, myocardial fibrosis, deep cytoplasmic staining, nuclear fission, and nuclear lysis, were also detected, together with an increase in the cardiac injury score, CK-MB and LDH enzymes and cTnI protein. These results indicate that MA can induce myocardial injury.
MA can induce oxidative stress and apoptosis. 33 Furthermore, dopamine overload, oxidative stress, mitochondrial dysfunction, endoplasmic reticulum stress, and protein degradation system dysfunction play important regulatory roles in MA-induced cardiotoxicity. 34 Our data showed that MDA content increased, SOD activity decreased, caspase-3 and Bax protein expression increased, and Bcl-2 expression decreased in the MA group, suggesting that MA may induce oxidative stress and apoptosis, leading to myocardial injury. Several studies have shown that MA can cause changes in Nrf2, 35 and Nrf2 exerts effects through its downstream antioxidant factor HO-1. 36 Therefore, we detected the protein expression levels of Nrf2 and HO-1 and found that both were increased in the MA group, 37 indicating that MA may activate the Nrf2/HO-1 pathway.
Based on our research, we found that MA induced myocardial injury, oxidative stress, and apoptosis, and activated the Nrf2/HO-1 signaling pathway. Nrf2 is an important transcription factor regulating oxidative stress. 38 In addition to maintaining redox homeostasis, Nrf2 also participates in various cellular processes, including myocardial cell apoptosis, thereby protecting the heart. 39 Therefore, we speculated that Nrf2 plays an important regulatory role in MA-induced cardiotoxicity. To test this, we explored the regulatory effects of Nrf2 on MA-induced cardiotoxicity via Nrf2 activation and Nrf2 knockout.
Several studies have reported that Nrf2 can protect the heart from damage, 39 as supported by our results. Notably, after Nrf2 activation by SFN, histopathological changes, cardiac injury score and cTnI protein expression levels were reduced compared with the MA group. Furthermore, these results were significantly increased after Nrf2 knockout compared with the MA group. These findings suggest that Nrf2 plays a protective role in MA-induced myocardial injury. Studies have also shown that Nrf2 has antioxidant effects. 20 Here, Nrf2 activation significantly decreased MDA content and significantly increased SOD activity compared with the MA group. In contrast, Nrf2 knockout significantly increased MDA content and significantly decreased SOD activity compared with the MA group. These results suggest that oxidative stress is more pronounced after knockout of the Nrf2 gene, and Nrf2 likely plays an antioxidant role in MA-induced oxidative stress in myocardial cells.
Activation of Nrf2 has been shown to suppress cardiomyocyte apoptosis in many disease models, and can inhibit ROCK2 expression,22,40 suggesting an interaction between Nrf2 and cardiomyocyte apoptosis. Several studies have suggested that activation of Nrf2 induces up-regulation of antioxidant target gene expression, leading to a decrease in intracellular oxidative stress, thereby inhibiting myocardial cell apoptosis.41,42 Nrf2 knockout mice are reported to show more severe phenotypes in disease models than wild-type mice,43,44 as found in the current study. Notably, knockout of Nrf2 aggravated the degree of myocardial cell apoptosis, while activation of Nrf2 attenuated the degree of myocardial cell apoptosis. However, the relationship between Nrf2 and apoptosis is complex. Various studies have suggested that Nrf2 affects apoptosis by affecting oxidative stress. ROS can activate apoptosis, however, when Nrf2 is activated, its downstream expression of related proteins, such as HO-1, can clear ROS, thereby reducing apoptosis.45,46 Thus, activation of Nrf2 may inhibit apoptosis of myocardial cells. In this experiment, compared with the MA group, Nrf2 activation decreased the protein expression levels of ROCK2, Caspase-3, and Bax in cardiomyocytes to varying degrees and increased the protein expression level of Bcl-2. These results suggest that Nrf2 activation attenuates the effects of apoptosis after MA administration. After Nrf2 knockout, the expression levels of these proteins showed the opposite trend, indicating that the degree of apoptosis was more obvious after Nrf2 knockout. Our results also demonstrated that Nrf2 inhibited the expression of ROCK2, although the specific mechanism remains unclear and requires further investigation. Overall, our findings strongly suggest that Nrf2 plays an important regulatory role in MA-induced apoptosis.
This study demonstrated the importance of Nrf2 in MA induced myocardial injury, Oxidative stress, and apoptosis. The cardiotoxicity was closely related to Nrf2 expression. However, the mutual influence between multiple results and the degree of impact did not discuss in this study, which have value for further research. After activating Nrf2, the degree of myocardial injury was reduced, but the therapeutic effect of the SFN was not clarified enough. The suitable drugs can activate Nrf2 and without side effect still need further search to discover.
Our study showed that MA-induced oxidative stress and apoptosis were inhibited by Nrf2 activation but aggravated by Nrf2 knockout (Figure 9). Furthermore, the Nrf2/HO-1 pathway attenuated MA-induced myocardial injury by regulating oxidative stress and apoptosis. In conclusion, our findings suggest that Nrf2 plays a key regulatory role in MA-induced cardiotoxicity and that MA can activate the Nrf2/HO-1 pathway to resist MA-induced myocardial injury, oxidative stress, and apoptosis. Our experiment not only explored cardiac damage induced by MA, but also highlighted the protective effects of the Nrf2/HO-1 signaling pathway. Our results should help to further explore MA toxicity, conducive to in-depth understanding of the cardiac damage induced by MA, and strengthen attention to the cardiotoxicity of MA abuser. The limitation of the present work was that it did not invovle deeply the potential mechanism of Nrf2. More evidences were needed to clarify the roles of Nrf2 in MA induced cardiotoxicity. Nrf2 attenuated MA-induced myocardial injury by regulating oxidative stress and apoptosis.
Footnotes
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the National Natural Science Foundation of China (No. 81860332 and No. 82260336); Yunnan Provincial Engineering Research Center of Forensic Medicine; Research Project of NHC Key Laboratory of Drug Addiction Medicine, Kunming Medical University (No. 2020DAMARC-002); Joint Research Project of Science and Technology Department of Yunnan Province & Kunming Medical University (No. 2019FE001(-163)); and Graduate Innovation Foundation of Kunming Medical University (No. 2022B03).