
Abstract
Sestrin2 (SESN2) is stress-inducible protein that confers cytoprotective effects against various noxious stimuli. Accumulating evidence has documented that SESN2 has potent anti-apoptosis and anti-oxidative stress functions. However, whether it provides neuroprotection in traumatic brain injury (TBI) models remains unexplored. The purpose of this study was to explore the regulatory effect of SESN2 on TBI using in vivo and in vitro models. We found that TBI resulted in a marked induction of SESN2 in the cerebral cortex tissues of mice. SESN2 overexpression in the brain by in vivo gene transfer significantly decreased neurological deficit, brain edema, and neuronal apoptosis of mice with TBI. Moreover, the overexpression of SESN2 significantly decreased the oxidative stress induced by TBI in mice. In vitro studies of TBI demonstrated that SESN2 overexpression decreased apoptosis and oxidative stress in scratch-injured cortical neurons. Notably, SESN2 overexpression increased the nuclear levels of nuclear factor-erythroid 2-related factor 2 (Nrf2) and enhanced the activation of Nrf2 antioxidant signaling in in vivo and in vitro models of TBI. In addition, the inhibition of Nrf2 significantly abolished SESN2-mediated neuroprotective effects in vivo and in vitro. In conclusion, these results of our work demonstrate that SESN2 protects against TBI by enhancing the activation of Nrf2 antioxidant signaling.
Introduction
Traumatic brain injury (TBI), causing motor dysfunction, neurological deficits and cognitive impairments, represents a primary cause of death and permanent disability in young adults. 1 Currently, TBI is a severe public health problem worldwide, causing heavy economic burdens to patients and their families. 2 However, there is still a shortage of effective therapeutic options for TBI. Mechanical injury to the head causes primary brain injury and subsequently induces secondary brain injury, which contributes to the development of TBI. 3 Oxidative stress has been acknowledged as a main contributor of secondary brain injury. 4 The excessive production of reactive oxygen species (ROS) induced by TBI results in genotoxicity, protein degradation, and lipid peroxidation, leading to neuronal apoptosis and brain damage. 4 The brain is an organ that is highly vulnerable to oxidative stress-induced damage. 5 Therefore, antioxidant therapy has been proposed as a promising therapeutic option for the treatment of TBI. 6
Sestrin2 (SESN2), a member of the Sestrin family, is a remarkable stress-inducible protein that is capable of providing cytoprotection against various noxious stimuli. 7 It is reported that SESN2 expression can be induced by hypoxia, ischemia, high glucose and endoplasmic reticulum. 8 –11 The dysregulation of SESN2 is implicated in various pathological conditions, including cancer, diabetes, aging and ischemia-related diseases. 12 Notably, SESN2 has been proposed as a potent antioxidant protein that strengthens the cellular antioxidant defense. 12 Currently, increasing studies have demonstrated that SESN2 exerts a neuroprotective function in various neurological disorders, including Parkinson’s disease and ischemic stroke. 13 –15
Nuclear factor-erythroid 2-related factor 2 (Nrf2) is physiologically critical for maintaining cellular redox homeostasis. 16 As a transcription factor, Nrf2 enters the nucleus and binds to antioxidant response elements (ARE) to promote the expression of antioxidant genes, such as heme oxygenase-1 (HO-1) and NAD(P)H: quinone oxidoreductase 1 (NQO-1). 17 Thus, the activation of Nrf2 antioxidant signaling can accelerate the clearance of ROS and relieve oxidative stress. Importantly, Nrf2 exerts a pivotal role in TBI. 18 Boosting Nrf2 antioxidant signaling provides remarkable neuroprotective effects against TBI-induced brain damage and neurological deficits. 19 –21 Therefore, Nrf2 is an interacting target for the treatment of TBI.
Although the neuroprotective role of SESN2 has been reported in different conditions, whether SESN2 exerts a role in TBI has not been well studied. The goals of this work were to assess whether SESN2 exerts a neuroprotective role in TBI using an experimental TBI model. We found that TBI treatment induced a significant increase in SESN2 expression in cerebral cortex tissues. The overexpression of SESN2 significantly decreased neurological deficit, brain edema, neuronal apoptosis and oxidative stress in TBI mice in vivo. In vitro studies of TBI demonstrated that SESN2 overexpression decreased the apoptosis and oxidative stress in scratch-injured cortical neurons. Further studies revealed that the up-regulation of SESN2 potentiated the activation of Nrf2 signaling. Notably, the inhibition of Nrf2 markedly abolished SESN2-mediated protective effects in TBI models. To summarize, our work demonstrates that SESN2 protects against TBI by enhancing the activation of Nrf2 antioxidant signaling.
Materials and methods
Animals and ethics statement
Male C57B6/J mice aged 8–10 weeks weighing 20–25 g purchased from Experimental Animal Center of Xi’an Jiaotong University Health Science Center (Xi’an, China) were utilized in this study. The mice were housed in a humidity-controlled environment at 23 ± 1°C on a 12 h light/dark cycle with access to food and water. The animal study was approved by the Ethics Committee of Xi’an Jiaotong University, and the experimental procedure was conducted in compliance with the principles of laboratory animal care and use approved by the Ethics Committee.
In vivo mouse model of TBI
The mouse model of TBI was established in accordance with a previously described protocol. 22 In brief, mice were anesthetized by intraperitoneal injection of 1% chloral hydrate (5 ml/kg), and placed in a stereotaxic frame. After mice were fixed on the platform, a midline longitudinal scalp incision was made to expose the skull. The left anterior frontal area was selected as the impact area, and a 200-g weight from a height of 2.5 cm free-fell onto the skull. The scalp wound was sutured and injured mice were returned to cages. Sham-injured mice subjected to same surgical procedures, but with no weight drop, were utilized as the control.
Intra-cerebroventricular injection
Recombinant adeno-associated virus expression SESN2 (AAV-SESN2) or lentivirus expressing SESN2 shRNA (LV-SESN2 shRNA) or Nrf2 shRNA (LV-Nrf2 shRNA) were synthesized by GenePharma (Shanghai, China). Mice were anesthetized and positioned in a stereotaxic frame. Then, a burr hole was drilled into the skull and recombinant viruses were injected using a Hamilton microsyringe through this hole. Thereafter, the scalp incision was sutured and the TBI model was established 3 days after intra-cerebroventricular injection.
Neurobehavioral evaluation
The neurological severity score (NSS) of mice was evaluated according to a modified score system that evaluated the motor, sensory, beam balance, reflex absence and abnormal movement functions of mice. Higher scores indicate that the neurological impairments are more serious.
Brain water content measurement
The brains of mice were dissected and weighed immediately to obtain the wet weight. Then, brains were dried for 3 days at 70°C to obtain dry weight. Brain water content was calculated according to the formula: (wet weight − dry weight)/(wet weight) × 100%.
Tissue collection and sectioning
Mice were euthanized and subjected to intracardiac perfusion with 0.9% saline through a thoracotomy via a cannula. The cortex tissues surrounding the contusion area was collected, frozen in liquid nitrogen and preserved at −80°C until use. The brain tissues were fixed in 4% paraformaldehyde and dehydrated in different concentrations of ethanol (45%, 55%, 65%, 75%, 85%, 95% and 100%). Then, the tissues were vitrificated by xylene, and made into paraffin blocks. The paraffin blocks were cut into 6 µm sections. The sections were dewaxed and boiled in a microwave with citrate buffer solution.
Real-time quantitative PCR (RT-qPCR)
The total RNA of cortex tissues or cultured neurons was isolated and purified by the RNeasy Mini Kit (QIAGEN, Hilden, Germany). First, the complementary DNA (cDNA) strand was generated by reverse-transcribing RNA using the FastKing-RT SuperMix and Enzyme (TianGen Biotech, Beijing, China). The levels of cDNA was quantified by RT-qPCR using FastFire qPCR PreMix (SYBR Green) (TianGen Biotech). The results of RT-qPCR were analyzed by the comparative threshold cycle (Ct) method to compute the relative expression of target genes, using β-actin for normalization.
Western blotting
Equal amounts of protein extracts were added to sodium dodecyl sulfate (SDS)-polyacrylamide gels for separation by electrophoresis. Then, electro-transfer experiments were performed to shift the proteins on SDS-polyacrylamide gels to Polyvinylidene Fluoride (PVDF) membranes using a Trans-Blot. PVDF membranes were blocked in 5% skimmed milk powder prior to primary antibody incubation. The primary antibodies were as follows: anti-SESN2, anti-Nrf2, anti-HO-1, anti-NQO-1, anti-β-actin, anti-Lamin B1 antibodies (Proteintech Group, Wuhan, China). After incubation with primary antibodies at 4°C overnight, membranes were incubated with horseradish peroxidase-conjugated IgG (Proteintech Group) and immuno-blotting bands were developed using a Genshare Enhanced Chemiluminescent Substrate Detection Ki (GseBio, Xi’an, China).
TUNEL assay
Apoptotic cells in the ipsilateral cortical cortex were evaluated by the TransDetect in situ Fluorescein TUNEL Cell Apoptosis Detection Kit (TransGen, Beijing, China). Briefly, the slices were incubated with Labeling Solution and TdT mixture for 1 h at 37°C in the dark. The slides were then washed and stained with DAPI for nucleus labeling. Then, fluorescence was observed and images were taken using a Leica fluorescence microscope.
Determination of malondialdehyde (MDA), superoxide dismutase (SOD) and glutathione peroxidase (GPx)
Tissue samples were homogenized in phosphate buffered saline and supernatants were collected by centrifugation. The protein concentrations were quantified with a BCA protein assay kit (Beyotime, Shanghai, China). The contents of MDA, SOD and GPx in the supernatants were measured by colorimetric method using commercial kits (Nanjing Jiancheng Biochemistry, Nanjing, China).
Isolation and culture of primary mouse cortical neurons
Cortical neurons were isolated from fetal mice brains and cultured in poly-D-lysine-coated dishes with neurobasal medium (Procell, Wuhan, China) supplemented with 1 mM glutamate, 2% B27 and 1% Penicillin & Streptomycin solution. Cells were maintained in a 5% CO2-containing atmosphere at 37°C. Half of the medium was replaced by new medium every 3 days. After culturing for 10–12 days, the cultured cortical neurons were subjected to in vitro studies.
In vitro model of TBI
In vitro traumatic injury was induced by manually scratching the confluent neuron cultures according to a previously described method. 23 Briefly, neurons were seeded into a six-well plate and manually scratched with a sterile plastic needle followed by a 9 × 9 square grid (with 4-mm spacing between the lines). Then, cultures were washed to remove cellular debris and scratched neurons were cultured for 24 h before detection. Un-scratched neurons served as a control.
Lactate dehydrogenase (LDH) assay
Cell injury of neurons was measured by LDH assay using LDH Cytotoxicity Assay Kit (Beyotime) according to the manufacturer’s protocols. Briefly, cells were treated with LDH release reagent and cultivated for 1 h at 37°C. The supernatants were collected by centrifugation and transferred to a 96-well plate at 120 μl/well. Then, 60 μl/LDH detection agent was added to each well and cultivated for 30 min in the dark at room temperature. Finally, the absorbance of the solution at 490 nm was assessed by an automatic ELISA analyzer.
Annexin V-FITC/PI flow cytometry assay
Neurons were collected, washed and resuspended in Annexin V-FITC binding buffer (Beyotime), followed by the addition of Annexin V-FITC reagent and PI reagent (Beyotime). Cells were gently mixed and cultivated for 10 min at room temperature in the dark. Then, samples were assessed by flow cytometry to determine the apoptotic rate.
Measurement of intracellular ROS level
Intracellular ROS levels were measured by a ROS sensitive probe 2′,7′-dichlorodihydrofluorescein diacetate (DCFH-DA). DCFH-DA is a non-fluorescent probe that is cable to entering into cells and is oxidized into fluorescent DCF. DCFH-DA was diluted into serum-free media to reach a final concentration of 10 μM. At the time of detection, cells were collected and resuspended in DCFH-DA-containing serum and cultivated for 30 min at 37°C in dark. Then, cells were washed and observed using a Leica fluorescence microscope, and images were taken.
Luciferase activity assay
The transcriptional activity of Nrf2/ARE was monitored using the pARE-luciferase reporter plasmid. Briefly, ARE-luciferase reporter plasmid and control Renilla luciferase vector were transfected into cells and cultured for indicated time. Thereafter, cells were harvested and lysed to measure luciferase activity within cells using Dual Luciferase Reporter Gene Assay Kit (Beyotime).
Statistical analysis
Experimental results were shown in the form of mean ± standard deviation. GraphPad Prism version 8.0 software was utilized to assess the group differences, using one-way analysis of variance followed by Tukey’s post-hoc test. The differences were deemed to be statistically significant when p < 0.05.
Results
SESN2 expression in the pericontusional cortex was induced by TBI in mice
Considering that SESN2 is a stress-inducible protein that exerts protective roles against various injuries, 7 we aimed to determine whether SESN2 expression is altered in response to TBI in a mouse model. Here, we found that SESN2 mRNA levels were significantly up-regulated in the pericontusional cortex 1 day after TBI compared with those in the sham group, and then gradually decreased at 3 and 7 days (Figure 1(a)). Similar to mRNA expression, protein expression of SESN2 was also increased by TBI compared with that in the sham group, as detected by Western blotting (Figure 1(b) and (c)). These data indicate that SESN2 expression is induced by TBI.

The effect of TBI on SESN2 expression in the pericontusional cortex of mice. (a) The effect of TBI on SESN2 mRNA expression in the pericontusional cortex of mice following injury at 1, 3 and 7 days was examined by RT-qPCR. (b, c) The effect of TBI on SESN2 protein expression in the pericontusional cortex of mice following injury at 1, 3 and 7 days was determined by Western blotting. *p < 0.05 and **p < 0.01 vs. sham.
Overexpression of SESN2 exerted neuroprotective effects against TBI in mice in vivo
To evaluate the role of SESN2 in TBI, we performed gain-of-function experiments of SESN2 in mice following TBI. The results showed that intra-cerebroventricular injection of AAV-SESN2 markedly up-regulated SESN2 expression in mice (Figure 2(a)–(c)). The results demonstrated that TBI significantly increased the NSS of mice, which was markedly decreased by SESN2 overexpression (Figure 2(d)). SESN2 overexpression significantly alleviated brain edema in mice with TBI (Figure 2(e)). Furthermore, TBI-induced neuronal apoptosis in the pericontusional cortex of mice was remarkably reduced by SESN2 overexpression (Figure 2(f), (g)). In addition, SESN2 overexpression ameliorated TBI-induced oxidative stress in the pericontusional cortex of mice (Figure 2(h)–(j)). Overall, these findings suggest that SESN2 exerts a neuroprotective role against TBI in mice in vivo.
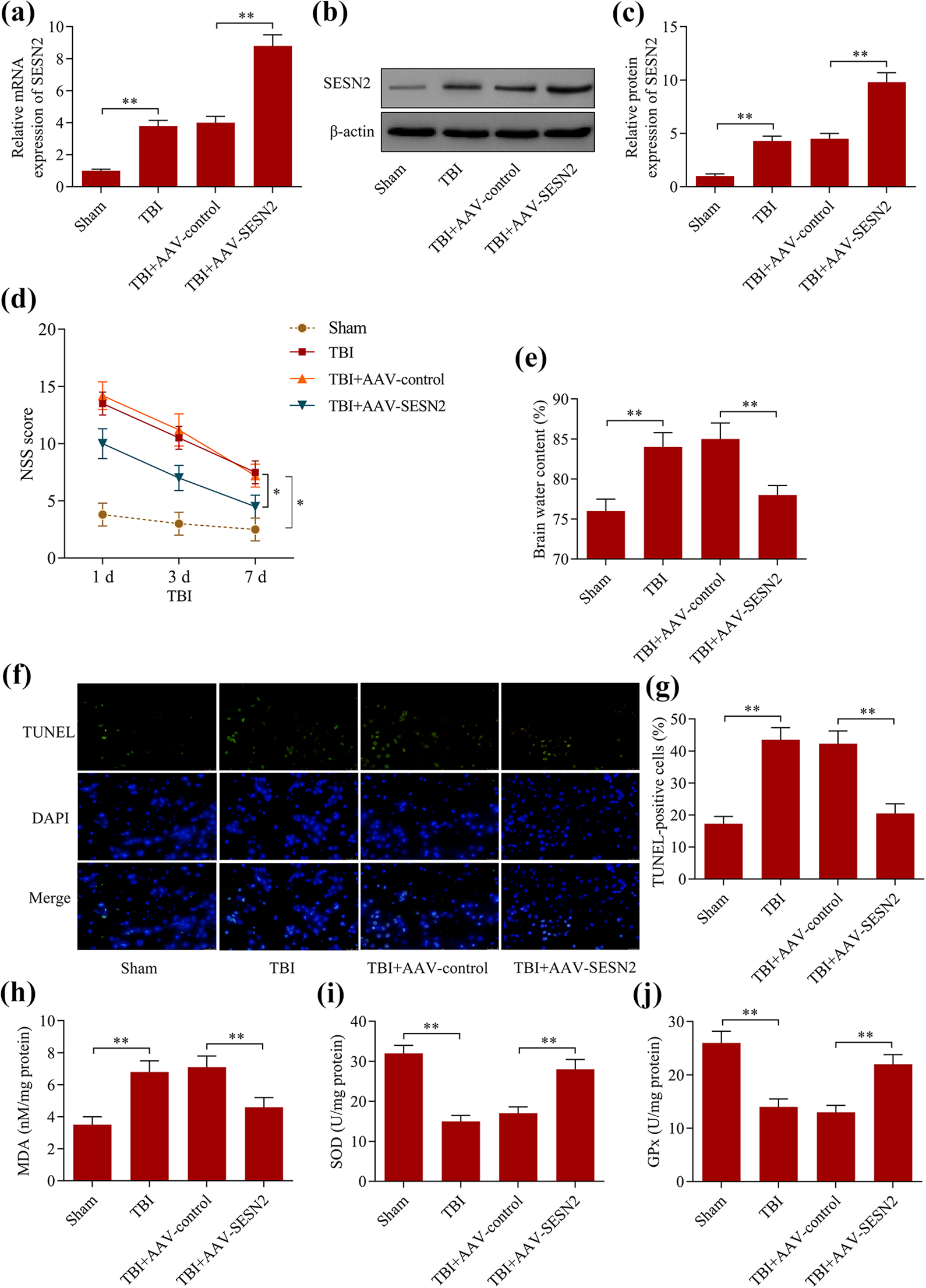
SESN2 overexpression provided neuroprotection after TBI in mice. AAV-SESN2 or AAV-control was intra-cerebroventricularly injected into mice prior to the establishment of TBI. (a) SESN2 mRNA levels were examined via RT-qPCR. (b, c) SESN2 protein levels were detected via Western blotting. (d) The effect of SESN2 overexpression on neurobehavioral changes of TBI mice was evaluated by the NSS test. (e) The effect of SESN2 overexpression on water content of TBI mice. (f, g) The effect of SESN2 overexpression on neuronal apoptosis in the pericontusional cortex of TBI mice was assessed by the TUNEL assay. The effect of SESN2 overexpression on the contents of (h) MDA, (i) SOD and (j) GPx in the pericontusional cortex of TBI mice was measured by ELISA kits. *p < 0.05 and **p < 0.01.
Loss of SESN2 exacerbated TBI-induced neurological dysfunction, apoptosis and oxidative stress
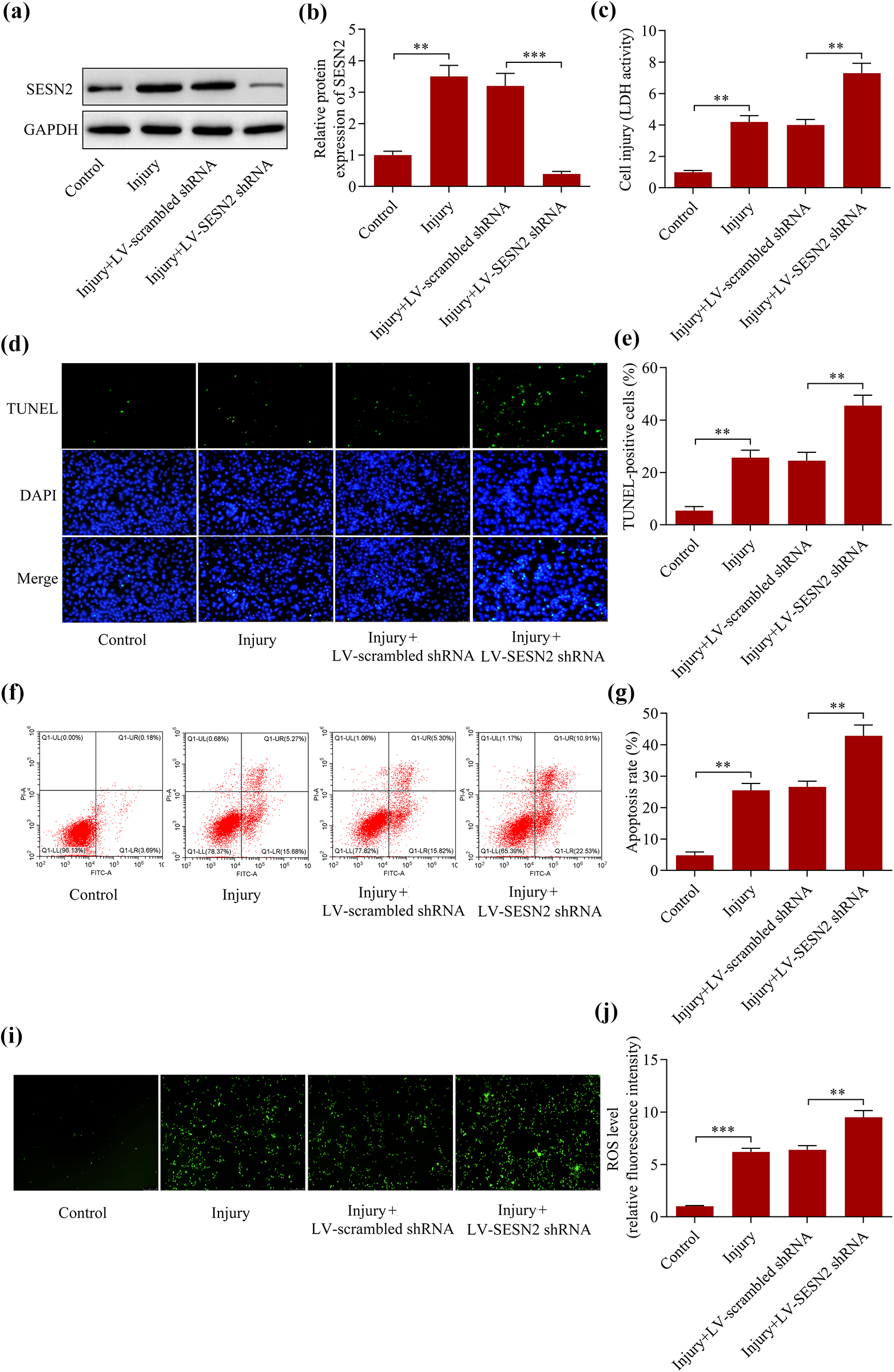
To further confirm the neuroprotective function of SESN2 in TBI mice, we next detected the effect of SESN2 loss on TBI-induced neurological dysfunction, apoptosis and oxidative stress. We found that the infection of LV-SESN2 shRNA into mice markedly depleted the expression of SESN2 (Figure 3(a)–(c)). The results demonstrated that the loss of SESN2 markedly aggravated the NSS of TBI mice (Figure 3(d)). The silencing of SESN2 significantly enhanced the brain edema of TBI mice (Figure 3(e)). Moreover, SESN2 knockdown exacerbated neuronal apoptosis (Figure 3(f) and (g)) and oxidative stress (Figure 3(h)–(j)). In summary, these data imply that the loss of SESN2 exacerbates TBI-induced neurological dysfunction, apoptosis and oxidative stress.

Loss of SESN2 exacerbated TBI-induced neurological dysfunction, apoptosis and oxidative stress. LV-SESN2 shRNA or LV-scrambled shRNA was intra-cerebroventricularly injected into mice prior to the establishment of TBI. (a) SESN2 mRNA levels were assessed via RT-qPCR. (b, c) SESN2 protein levels were examined via Western blotting. (d) The effect of SESN2 silencing on neurobehavioral changes of TBI mice was assessed via the NSS test. (e) The effect of SESN2 silencing on the water content of TBI mice. (f, g) The effect of SESN2 silencing on neuronal apoptosis in the pericontusional cortex of TBI mice was detected via the TUNEL assay. The effect of SESN2 silencing on the contents of (h) MDA, (i) SOD and (j) GPx in the pericontusional cortex of TBI mice was monitored via ELISA kits. *p < 0.05, **p < 0.01 and ***p < 0.001.
Overexpression of SESN2 protects cultured neurons against scratch-induced injury
To validate the neuroprotective role of SESN2 in TBI, we further investigated the regulatory effect of SESN2 using an in vitro model of TBI, which is established by scratching the primary mouse cortical neurons. We found that SESN2 expression was also induced by scratch in cultured neurons (Figure 4(a) and (b)). As expected, we found that SESN2 overexpression significantly suppressed the scratch-induced injury of cultured neurons (Figure 4(c)). Moreover, scratch-induced apoptosis of cultured neurons was markedly decreased by SESN2 overexpression (Figure 4(d)–(g)). In addition, overexpression of SESN2 also remarkably decreased the ROS production in scratched neurons (Figure 4(h) and (i)). On the contrary, SESN2 silencing enhanced the cultured neurons to scratch-induced injury (Figure 5(a)–(i)). Overall, these results confirm that SESN2 exerts a neuroprotective role in an in vitro model of TBI.

Overexpression of SESN2 ameliorated scratch-induced injury of cultured neurons. Primary mouse cortical neurons were infected with AAV-SESN2 or AAV-control prior to scratch injury. (a, b) Protein levels of SESN2 were assessed via Western blotting. (c) The effect of SESN2 overexpression on scratch-induced neuronal injury was tested via the LDH assay. The effect of SESN2 overexpression on scratch-induced neuronal apoptosis was measured by (d, e) TUNEL assay and (f, g) Annexin V-FITC/PI assay. (h, i) The effect of SESN2 overexpression on scratch-induced ROS generation was monitored by ROS assay. **p < 0.01 and ***p < 0.001.
SESN2 silencing enhanced the cultured neurons to scratch-induced injury. Primary mouse cortical neurons were infected with LV-SESN2 shRNA or LV-scrambled shRNA prior to scratch injury. (a, b) protein levels of SESN2 were determined via Western blotting. (c) The effect of SESN2 silencing on scratch-induced neuronal injury was assessed via LDH assay. The effect of SESN2 silencing on scratch-induced neuronal apoptosis was evaluated via (d, e) the TUNEL assay and (f, g) the Annexin V-FITC/PI assay. (h, i) The effect of SESN2 silencing on scratch-induced ROS generation was evaluated via ROS assay. **p < 0.01 and ***p < 0.001.
SESN2 enhanced Nrf2 activation in TBI mice in vivo
Previous studies have revealed that SESN2 exerts the protective role associated with the up-regulation of Nrf2 antioxidant signaling. 15,24,25 Considering that Nrf2 also plays a key role in TBI, we sought to determine whether SESN2 regulates Nrf2 signaling in TBI. Here, we found that the overexpression of SESN2 markedly increased the protein levels of nuclear Nrf2 (Figure 6(a) and (b)), while it decreased the protein levels of cytoplasmic Nrf2 (Figure 6(c) and (d)) in the pericontusional cortex of TBI mice. Furthermore, SESN2 overexpression significantly promoted the expression of Nrf2 target genes, HO-1 and NQO-1 (Figure 6(e)–(g)). On the contrary, loss of SESN2 significantly impeded the activation of Nrf2 signaling in the pericontusional cortex of TBI mice (Figure 7(a)–(g)). In summary, these data suggest that SESN2 contributes to Nrf2 activation in TBI mice.

Overexpression of SESN2 potentiated Nrf2 activation in TBI mice. The effect of SESN2 overexpression on protein levels of (a, b) nuclear or (c, d) cytoplasmic Nrf2 was measured via Western blotting. The effect of SESN2 overexpression on mRNA levels of (e) HO-1 and (f) NQO-1 was determined via RT-qPCR. (g) The effect of SESN2 overexpression on protein levels of HO-1 and NQO-1 was determined via Western blotting. **p < 0.01.

Loss of SESN2 impeded Nrf2 activation in TBI mice. The effect of SESN2 silencing on protein levels of (a, b) nuclear or (c, d) cytoplasmic Nrf2 was assessed by Western blotting. The effect of SESN2 silencing on mRNA levels of (e) HO-1 and (f) NQO-1 was examined by RT-qPCR. (g) The effect of SESN2 silencing on protein levels of HO-1 and NQO-1 was evaluated by Western blotting. **p < 0.01.
SESN2 promoted Nrf2 activation in scratch-injured neurons in vitro
To verify whether SESN2 regulates Nrf2 signaling in TBI, we further investigated the effect of SESN2 on Nrf2 signaling in scratch-injured neurons in vitro. The results demonstrated that SESN2 overexpression markedly increased the protein levels of nuclear Nrf2 (Figure 8(a) and (b)), while it decreased the protein levels of cytoplasmic Nrf2 (Figure 8(c) and (d)) in scratch-injured neurons. Moreover, SESN2 overexpression remarkably up-regulated the transcriptional activity of Nrf2 (Figure 8(e)), and increased the expression of Nrf2 target genes (Figure 8(f)). On the contrary, knockdown of SESN2 repressed the activation of Nrf2 signaling in scratch-injured neurons (Figure 8(g)–(l)). Collectively, these data confirm that SESN2 promoted Nrf2 activation in scratch-injured neurons and in vitro.

SESN2 promoted Nrf2 activation in scratch-injured neurons in vitro. The effect of SESN2 overexpression on protein levels of (a, b) nuclear or (c, d) cytoplasmic Nrf2 was examined by Western blotting. (e) The effect of SESN2 overexpression on Nrf2 transcriptional activity was monitored by the ARE-dependent luciferase reporter assay. (f) The effect of SESN2 overexpression on the protein levels of HO-1 and NQO-1 was measured by Western blotting. The effect of SESN2 knockdown on the protein levels of (g, h) nuclear or (i, j) cytoplasmic Nrf2 was determined by Western blotting. (k) The effect of SESN2 knockdown on Nrf2 transcriptional activity was monitored by the ARE-dependent luciferase reporter assay. (l) The effect of SESN2 knockdown on the protein levels of HO-1 and NQO-1 was assessed by Western blotting. **p < 0.01.
Inhibition of Nrf2 abolished SESN2-overexpression-mediated neuroprotective effect in TBI mice in vivo
To determine whether Nrf2 contributes to SESN2-mediated neuroprotection in TBI mice, we further investigated the effect of Nrf2 inhibition on SESN2-overexpression-mediated effects. The results showed that the infection of LV-Nrf2 shRNA significantly down-regulated the levels of nuclear Nrf2 (Figure 9(a)), and reversed the promotion effect of SESN2 overexpression on the expression of Nrf2 target genes (Figure 9(b)). The suppressive effect of SESN2 overexpression on NSS (Figure 9(c)) brain edema (Figure 9(d)) in TBI mice was also abolished by Nrf2 inhibition. In addition, Nrf2 inhibition also reversed the suppressive effect of SESN2 overexpression on neuronal apoptosis (Figure 10(a) and (b)) and oxidative stress (Figure 10(c)–(e)) in TBI mice. To summarize, these data suggest that SESN2 overexpression provides neuroprotection in TBI mice by enhancing Nrf2 signaling.

SESN2 overexpression provides neuroprotection in TBI mice via enhancing Nrf2 signaling. AAV-SESN2 or/and LV-shRNA was intra-cerebroventricularly injected into mice prior to the establishment of TBI. (a, b) Protein levels of nuclear Nrf2, HO-1 and NQO-1 were detected via Western blotting. (c) Neurobehavioral changes of TBI mice was evaluated by the NSS test. (d) Water content of TBI mice. *p < 0.05 and **p < 0.01.

Nrf2 inhibition reversed the SESN2-mediated suppressive effect on TBI-induced neuronal apoptosis and oxidative stress. (a, b) Neuronal apoptosis in the pericontusional cortex of TBI mice was assessed by TUNEL assay. the contents of MDA (c), (d) SOD and (e) GPx in the pericontusional cortex of TBI mice was measured by ELISA kits. **p < 0.01.
Suppression of the Nrf2 reversed SESN2-overexpression-mediated neuroprotective effect in scratch-injured neurons in vitro
To further confirm whether SESN2 exerts the neuroprotective function via Nrf2, we assessed the effect of Nrf2 inhibition on SESN2-overexpression-mediated effects in scratch-injured neurons in vitro. Here we found that the infection of LV-Nrf2 shRNA markedly decreased the levels of nuclear Nrf2 in scratch-injured neurons (Figure 11(a) and (b)), and reversed the promotion effect of SESN2 overexpression on the activation of Nrf2 signaling (Figure 11(c)). In addition, the suppressive effect of SESN2 overexpression on scratch-induced apoptosis (Figure 11(d) and (e)), and ROS production (Figure 11(f) and (g)) was markedly abolished by Nrf2 inhibition. Overall, these results confirm that SESN2 exerts neuroprotective effects through enhancing Nrf2 signaling.

Suppression of Nrf2 reversed SESN2-overexpression-mediated neuroprotective effects in scratch-injured neurons. Cortical neurons were infected with AAV-SESN2 or/and LV-Nrf2 shRNA prior to scratch injury. (a, b) Protein levels of nuclear Nrf2, HO-1 and NQO-1 were assessed via Western blotting. (c) Nrf2 transcriptional activity was monitored via the ARE-dependent luciferase reporter assay. (d, e) Neuronal apoptosis was evaluated by the Annexin V-FITC/PI assay. (f, g) Intracellular levels of ROS were measured by the ROS assay. **p < 0.01 and ***p < 0.001.
Discussion
This work for the first time reveals a neuroprotective role of SESN2 in TBI. Our data demonstrated that SESN2 was induced by TBI and that SESN2 up-regulation attenuated the neurological deficit, brain edema, neuronal apoptosis and oxidative stress seen in TBI mice. Moreover, SESN2 overexpression exerts a neuroprotective effect in scratch-injured neurons in vitro. Notably, we identified that the neuroprotective role of SESN2 in TBI was associated with its enhancing effect on the activation of Nrf2 antioxidant signaling (Figure 12). Therefore, our work underlines a pivotal role of the SESN2/Nrf2 axis in the development of TBI.

A schematic diagram of SESN2/Nrf2 axis in protection against TBI.
SESN2 is a remarkable stress-inducible protein that enables various types of cells to survive under diverse noxious stimuli. 7,11,26 –28 In particular, Sesn2 also exerts a cytoprotective role in neurons in response to noxious stimuli by down-regulating cell apoptosis and oxidative stress. The up-regulation of SESN2 represses rotenone-induced apoptosis in dopaminergic neurons. 29 SESN2 expression is induced by the exposure of amyloid β-peptide in cortical neurons, and SESN2 silencing aggravates amyloid β-peptide-induced neuronal injury and oxidative stress. 30 The overexpression of SESN2 ameliorates hydrogen peroxide-induced apoptosis and oxidative stress in retinal ganglion cells. 24 Moreover, the up-regulation of SESN2 protects neurons from oxygen-glucose deprivation/re-oxygenation-induced apoptosis and oxidative stress in vitro. 15,25 These studies suggest a considerable neuroprotective function of SESN2. Consistently, in this study, we found that SESN2 expression was induced in cortical neurons injured by scratch, an in vitro model of TBI. Notably, the up-regulation of SESN2 alleviated the apoptosis and ROS generation in scratch-injured neurons. Therefore, our work confirms that SESN2 also exerts a protective role against scratch-induced neuronal injury.
SESN2 is extensively implicated in the pathogenesis of neurological disorders. SESN2 participates in the pathophysiology of human immunodeficiency virus-associated neurocognitive disorders. 31 The induction of SESN2 provides a protective effect in Parkinson’s disease via suppressing oxidative stress. 13 SESN2 expression is induced by ischemic injury in hippocampal regions of brains. 32 Notably, overexpression of SESN2 ameliorates cerebral ischemia/reperfusion injury via suppressing oxidative stress and increasing angiogenesis. 15,33 Considering these outstanding neuroprotective effects of SESN2 in vivo, we speculated that SESN2 may also provide neuroprotection against TBI. As expected, we found that SESN2 expression was induced by TBI in the pericontusional cortex of mice. Notably, the overexpression of SESN2 in the mouse brain by gene transfer markedly ameliorated TBI-induced neurological deficit, brain edema, neuronal apoptosis and oxidative stress in mice. In addition, the silencing of SESN2 made the mice more vulnerable to TBI-induced brain damage. Thus, the findings of our study indicate that SESN2 plays a crucial role in the development of TBI, and serves as a promising target for the treatment of TBI.
Nrf2-mediated antioxidant signaling plays a crucial role in TBI. 18,34 Nrf2 activity is increased after TBI, which aims to boost the antioxidant defense to provide neuroprotective effects. 35,36 Targeting Nrf2 has been suggested as a promising strategy for the treatment of TBI. 20,37,38 Interestingly, SESN2 has emerged as a positive regulator of Nrf2. 7,39,40 It is reported that SESN2 promotes Nrf2 activation by down-regulating the Nrf2 repressor, Keap1. 24,26,41 In addition, SESN2 is also reported to enhance Nrf2 activation via regulating adenosine monophosphate activated protein kinase. 42 In general, SESN2 acts as a pivotal modulator of Nrf2 antioxidant signaling. Consistently, the findings of our work demonstrated that SESN2 also contributed to Nrf2 activation in TBI. Our data showed that the up-regulation of SESN2 increased the nuclear translocation of Nrf2 and promoted the transcription of Nrf2 target genes via in vivo and in vitro models of TBI. Moreover, we identified that the inhibition of Nrf2 markedly reversed SESN2-overexpression-mediated neuroprotective effects in TBI models. Collectively, our work shows that SESN2 protects from TBI by potentiating the activation of Nrf2-mediated antioxidant signaling.
This work demonstrates that SESN2 exerts a neuroprotective role against TBI by relieving neurological deficit, brain edema, and neuronal apoptosis by enhancing the activation of Nrf2 signaling. Our study underlines a pivotal role of SESN2/Nrf2 signaling in the development of TBI. Targeting SESN2 may provide novel therapeutic options for the treatment of TBI.
Footnotes
Author contributions
XL and ML designed the study, performed the experiments, and drafted the manuscript. JZ and WH analyzed the data. JS designed the study, supervised the study, and revised the manuscript.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.