
Abstract
Riboflavin deficiency produces severe peripheral neve demyelination in young, rapidly growing chickens. While this naturally-occurring vitamin B2 deficiency can cause a debilitating peripheral neuropathy, and mortality, in poultry flocks, it can also be a useful experimental animal model to study the pathogenesis of reliably reproducible peripheral nerve demyelination. Moreover, restitution of normal riboflavin levels in deficient birds results in brisk remyelination. It is the only acquired, primary, demyelinating tomaculous neuropathy described to date in animals. The only other substance that causes peripheral nerve demyelination similar to avian riboflavin deficiency is tellurium and the pathologic features of the peripheral neuropathy produced by this developmental neurotoxin in weanling rats are also described.
Introduction
Peripheral neuropathies are one of the most common neurologic disorders and of worldwide distribution. Peripheral nerve disorders can be classified according to the structural element principally injured, namely neuronopathies, axonopathies (axonal degeneration, with preservation of the nerve cell body), myelinopathies (involving direct damage to the Schwann cell or myelin, leading to segmental demyelination, with relative preservation of axonal integrity), and injury to the nerve-associated vasculature and connective tissue, with secondary bystander damage to nerve fibres. 1 Segmental demyelination is the major lesion of primary myelinopathy in the peripheral nervous system, one or several internodes of the myelin sheath being injured, with relative preservation of the axon. 1 This classification also recognizes the functional interdependence of these different constituents of the peripheral nerve. Peripheral neuropathies can also be categorized according to their spatial distribution (diffuse or multifocal, symmetrical or asymmetrical) and time course (acute, subacute, or chronic) 2 or etiology (genetic, immune, infectious, metabolic, nutritional, toxic, traumatic, or idiopathic). 3 Immune-mediated insults to nerve fibres, and infectious neuropathies, are associated with inflammation, the latter group due to innate and acquired immune responses to microbial infections. In humans, metabolic cases are most commonly produced by diabetes mellitus, while toxic injury is frequently induced by chemotherapeutic agents.2,3
Apart from autoimmune neuropathies, there are very few effective therapies that specifically target the underlying pathogenic mechanisms of axonal degeneration (axonopathies) or Schwann cell dysfunction (myelinopathies). Treatments are usually directed at symptoms and of variable benefit. Accordingly, the development of effective mechanism-based therapeutic interventions requires animal models that replicate the essential morphologic features of a particular peripheral neuropathy in order to facilitate translatability in human clinical trials.
Although the pathogenesis of demyelination, and remyelination, in peripheral nerves is still incompletely understood, only a small group of non-immunologic animal models have been developed to study these pathologic processes. Weanling rats fed a diet containing elemental tellurium develop a highly synchronized, but transient, demyelination of peripheral nerves, which is closely followed by Schwann cell proliferation and rapid remyelination. 4 Similarly, in young chickens, a deficiency of riboflavin (vitamin B2) produces demyelination in peripheral nerves and, when this vitamin deficiency is reversed, there is rapid remyelination. 5 In other animal models, such as lead intoxication, there is asynchronous demyelination and remyelination, while topical application of lysolecithin and diphtheria toxin cause a more localized demyelination of peripheral nerve fibres. 2
This review will outline the morphologic features of the demyelination, and remyelination, produced in peripheral nerves of chickens with riboflavin deficiency and highlight the novel insights this model has provided into these two processes. It also describes a similar peripheral nerve demyelination produced by tellurium in weanling rats.
Riboflavin
Riboflavin (7,8-dimethyl-10-ribityl-isoalloxa zine), also known as vitamin B2, is one of the B vitamins, all of which are water-soluble. It is an essential dietary nutrient since higher species have lost the ability to synthesize this vitamin. There is, however, a limited endogenous synthesis of riboflavin by large intestinal bacteria.6–9
The most important biologically active forms of riboflavin are flavin adenine dinucleotide (FAD) and flavin mononucleotide (FMN). They comprise more than 90% of dietary riboflavin. Riboflavin has a central role as a redox coenzyme in energy-yielding metabolism. The metabolic function of the flavin coenzymes is as electron carriers in a wide range of oxidation and reduction reactions essential for all metabolic processes, including the electron transport chain. They also have a role in chromatin remodelling, DNA repair, protein folding, and apoptosis.8–10 Riboflavin has an important role in myelin formation, selectively intervening in peripheral nerve myelin synthesis. In young chickens with riboflavin deficiency, significant components of the myelin membrane, including myelin lipids, cerebrosides, phosphatidylethanolamine, and sphingomyelin, are depleted.8,9 Absorption of this vitamin is limited and any absorbed in excess of requirements is rapidly excreted in urine. However, there is very efficient conservation and reutilization of this vitamin in tissues, but no evidence of significant storage in liver, heart and kidneys. 10
Apart from milk and eggs, which contain significant amounts of protein-bound riboflavin, most of this vitamin in foods occurs as flavin coenzymes, especially FAD, bound to enzymes. After absorption from the proximal small intestine, tissue-specific transporter proteins direct riboflavin to the intracellular machinery, especially for the biosynthesis of the flavocoenzymes, FAD and FMN, and human riboflavin transporter deficiency disorders (Brown-Vialetto-Van Laere and Fazio-Londe diseases) are autosomal recessive conditions caused by mutations of genes encoding riboflavin transporters.8–10 Since reoxidation of reduced flavin coenzymes is the principal source of oxygen radicals in the body, there is very strict control of riboflavin content in the human body. 10
Riboflavin is vital for the proper functioning of the nervous, but also endocrine, cardiovascular, and immune, systems. Riboflavin deficiency has also been implicated as a risk factor for multiple sclerosis and has been proposed as a putative neuroprotective agent for this demyelinating disease.11,12
Naturally-occurring riboflavin deficiency
Although riboflavin is poorly stored in humans, a pure deficiency (ariboflavinosis) is rare in Western countries due to its presence in amounts exceeding normal requirements in many foods. When this deficiency does occur, for example in malabsorption syndromes, riboflavin deficiency is generally associated with a multi-vitamin deficiency, and the signs and symptoms manifest partly reflect these other deficiencies. There is no well-defined riboflavin deficiency disease and, while the early signs of riboflavin deficiency are readily reversible, supplementation rarely reverses later pathologic changes. Riboflavin deficiency is, nevertheless, endemic in some, particularly developing, countries, due to a low dietary intake. The main effect of riboflavin deficiency is on lipid metabolism and this deficiency can impair the metabolism of other nutrients, particularly other B vitamins, by diminished levels of flavin coenzymes. Riboflavin deficiency in humans is signaled by glossitis, cheilosis, dermatitis, anemia, and neuropathy.8–10
In domestic animal species, riboflavin deficiency mainly occurs in pigs and chickens fed grain rations with suboptimal concentrations of this vitamin. Ruminants do not become deficient because of rumen synthesis of B-complex vitamins, although young calves may develop this deficiency if deprived of milk or an appropriate milk replacer.5,13
Riboflavin is one of the vitamins likely to be deficient in commercial chicken feeds and the nutritional requirements for this vitamin vary, depending on factors such as genetics, stage of growth, and environmental conditions. The requirements for riboflavin are highest for newly hatched chicks and chickens used for breeding. The most frequent cause of riboflavin deficiency in poultry is inadvertent omission of a complete vitamin premix in the diet, but vitamin B2 is can also be degraded by light and pH, and feed should not be stored for greater than 2 months. When a diet is unintentionally devoid of all vitamins, it is the clinical signs of riboflavin deficiency that appear first, a deficiency of body stores of fat-soluble vitamins taking longer to become manifest.5,13,14
When chicks are fed a diet deficient in riboflavin, they grow slowly and become weak and emaciated, but are not inappetent. Diarrhea develops between one and 2 weeks. Deficient chicks are reluctant to move unless driven, and frequently walk on their hocks with the aid of their wings. The toes on one or both feet are curled under and flexed, giving rise to the designation of this vitamin deficiency as “curled toe paralysis”. In advanced stages of the disease, chicks tend to be prostrate with drooping wings and legs are splayed out in different directions. Leg muscles are atrophic and flabby and the skin is dry and harsh.5,13,14
If the riboflavin deficiency is low-grade, clinical signs are non-specific. However, the presence of “curled toe paralysis” is regarded as diagnostic, supported by ancillary tests such as histopathology of peripheral nerves, a therapeutic response to vitamin B2 administration, and assay of the suspected deficient feed. The principal differential diagnosis is Marek’s disease, which is caused by a herpesvirus, and results in neoplastic infiltration and enlargement of peripheral nerves by T-cell lymphoma (neurolymphomatosis).5,12,13
Treatment and prevention of riboflavin deficiency relies on an adequate supply of this vitamin. Two, 100 µm doses of riboflavin will correct this deficiency in chickens, followed by inclusion of an adequate level in the ration. However, when the deficiency is longstanding, there is irreparable damage to peripheral nerves, and administration of riboflavin is not curative. Chickens given diets which are only partially riboflavin deficient may recover spontaneously, since the requirement for vitamin B2 decreases with age.5,13,14
A dietary riboflavin deficiency in hens results in decreased egg production, increased embryonic mortality, and hepatomegaly due to fatty change (steatosis). The hatchability of eggs declines, but can return to normal within a week when the diet becomes again vitamin B2 replete. Embryos that fail to hatch often have defective down (termed “clubbed down”), which is a failure of the down feather follicle to rupture the surrounding sheath, causing the feather to assume a coiled appearance. “Clubbed down” is an important diagnostic indicator of riboflavin deficiency in chickens.5,13,14
At autopsy, peripheral nerves appear grossly enlarged and, microscopically, there is a segmental demyelinating neuropathy, which is attended by endoneurial oedema, but only mild, patchy axonal degeneration. Affected Schwann cells are hypertrophied and contain myelin debris and many lipid droplets, but macrophage involvement has not been properly assessed. Remyelination is correlated temporally with clinical improvement and is attributed to a declining riboflavin requirement with increasing age, and decreasing growth rate, coupled with an age-associated increase in intestinal synthesis of this vitamin. A lack of riboflavin-derived coenzymes, FAD and FMN, has been postulated as the basis for Schwann cell dysfunction and resulting demyelination, an impairment of oxidative phosphorylation leading to diminished cellular energy levels in Schwann cells at a time of rapid growth and maximal metabolic demand in affected birds.5,13
Riboflavin deficient animals have a lower metabolic rate than vitamin-replete controls and require a 15%–20% higher food intake to maintain body weight. In normal laying hens, induction of a riboflavin-binding protein results in a 100-fold increase in plasma riboflavin, compared with non-laying females or males. Moreover, in mutant chickens lacking this protein, there is a massive urinary loss of riboflavin and, although embryos develop normally for about 10 days, they then develop a severe hypoglycemia. 10
With respect to the prevention of vitamin deficiencies in humans, poultry meat, in addition to being a high quality protein, is an important and rich source of highly bioavailable micronutrients, including vitamins such as riboflavin. By contrast, these micronutrients are either not present in plant foods or have low bioavailability. 7
Experimental riboflavin deficiency
Experimental studies have been conducted on avian riboflavin deficiency by three different groups to examine the processes of demyelination and remyelination in peripheral nerves. In the first study (Figure 1), newborn, rapidly growing broiler meat chickens were fed either a riboflavin deficient diet containing 1.8 mg/kg riboflavin or a conventional diet containing 5.0 mg/kg riboflavin.15–18 The normal riboflavin requirement for rapidly growing, meat-type chickens is 3.6 mg/kg of feed. 19 Liver riboflavin levels were analyzed on postnatal day 11 (PN 11) by a microbiologic method using Lactobacillus casei as the test organism, the growth of this riboflavin-dependent bacterium correlating with the amount of this vitamin in the sample. Peripheral nerves were fixed by transcardiac perfusion with 4% paraformaldehyde/2.5% glutaraldehyde in 0.1 M phosphate buffer (pH 7.4) on PN 6, 11, 16 and 21. Sciatic and brachial nerves were collected and processed for light and electron microscopic examination and teased nerve fibre studies. Control birds fed the conventional diet were killed at each of these time-points.
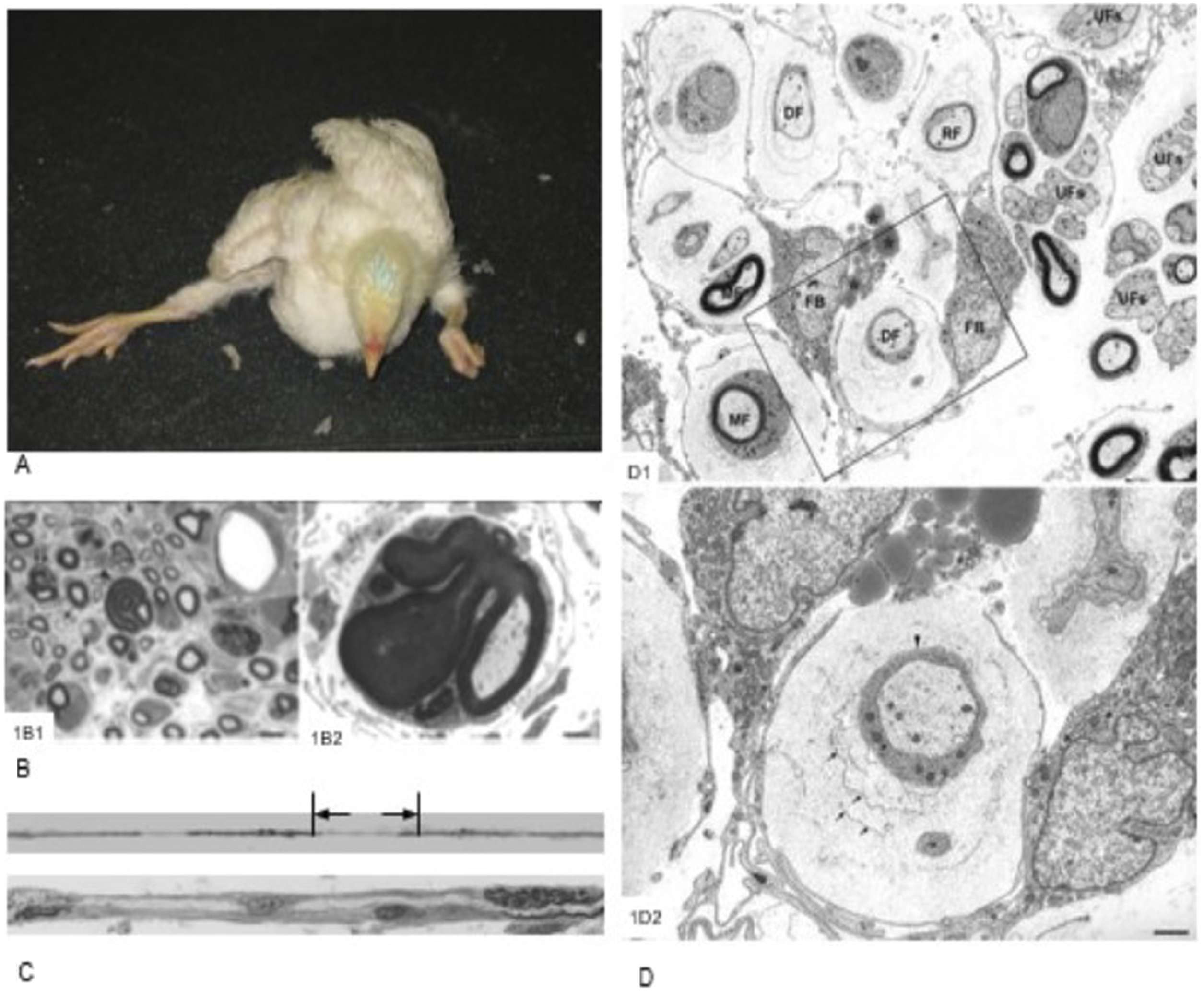
Clinicopathologic findings in riboflavin deficient chickens. (1a): A 21-day-old, riboflavin-deficient chicken presents in sternal recumbency, with the legs splayed and toes curled. (1b–c): Pathologic findings in a 16-day-old, riboflavin-deficient chicken. (1b1). A plastic section stained with toluidine blue shows endoneurial edema, active myelin degeneration, a naked axon (arrows), due to complete demyelination, and a tomaculum resulting from redundant myelin folding. Bar = 10 µm. (1b2). Electron micrograph showing a tomaculum with redundant myelin folding. Bar = 2 µm. (1c). Upper panel shows part of a teased nerve fiber with segmental demyelination and paranodal swellings. The lower panel shows a longitudinal section through the designated site, showing a naked axon with attached fibroblasts and active myelin degeneration. (1d). Electron micrograph from the sciatic nerve of a 31-day-old, riboflavin-deficient chicken showing cytoplasmic processes from two fibroblasts (FB) surrounding myelinated (MF), demyelinated (DF), and remyelinating (RF) fibers. Rough endoplasmic reticulum and lipid droplets (asterisk) are present in the fibroblast cytoplasm. The neighboring unmyelinated fibres (UFs), together with corresponding Schwann cells, are normal. Higher magnification demonstrates a demyelinated axon enwrapped by a Schwann cell with basement membrane (arrowhead) surrounded by cytoplasmic processes from two fibroblasts. The space enclosed by the fibroblast cytoplasmic processes contains negatively stained collagen fibrils and concentric fibroblast plasma membranes. Bar = 1 µm. 1b and 1d are adapted from Cai et al (2007, 2009) with permission from publishers and authors.
Neurologic signs were observed from PN8, affected chickens initially being incoordinated and, although alert, were reluctant to move. With progressively more severe paresis, birds were unable to extend their hocks and toes tended to be flexed and curled under. With increasing time, stunting occurred as birds were either reluctant, or unable, to access feed. No neurologic signs were found in control birds. The riboflavin concentration in birds on a deficient diet (mean 24 µg/g) was significantly lower (p < .01) than control birds fed a conventional diet (32 µg/g).15,16,18
Tomacula were detected early during demyelination on PN 11 and became more numerous and prominent with increasing time. Tomacula are sausage-shaped (from the Latin tomaculum meaning sausage) overgrowths and expansions of the myelin sheath due to redundant myelin folding. These focal myelin swellings were characterized by complex wrapping of redundant myelin loops around the nerve fibre. From PN 16 onwards, some tomacula showed myelin splitting and degeneration and appeared to be unstable. In teased nerve fibres examined at PN 11, tomacula were found to be located at the paranodal region and, by PN 16 and PN 21, were attended by segmental demyelination involving the internodal region. Regeneration of myelin was detected at PN 16 and, at PN 21, remyelination was prominent. Axonal degeneration was minimal, but Schwann cells were hypertrophied and contained myelin debris and numerous lipid droplets.15,16,18
Avian riboflavin deficiency appears to be the first animal disease in which an acquired, primary, demyelinating tomaculous neuropathy has been reported, although a tomaculous neuropathy of suspected hereditary origin has been reported in cattle 20 and dogs. 21 In humans, various demyelinating neuropathies, including different types of Charcot-Marie-Tooth disease and chronic inflammatory demyelinating polyneuropathy (CIDP), are associated with tomacula formation in peripheral nerves. 1 The term “tomaculous neuropathy” classically refers to hereditary neuropathy with liability to pressure palsies (HNPP) caused by a mutation in the gene coding for the peripheral myelin protein 22 (PMP22), but the term should strictly be used only in a descriptive sense. 1 Although tomacula are generally considered to be associated with remyelination, the finding of tomacula early in the paranodal region of otherwise normal myelinated nerve fibres in the present study suggested that degeneration of tomacula led to segmental demyelination. This myelin loss occurred initially paranodally, then progressed to involve the internodal region, an internode covering the length of an axon between two nodes of Ranvier and constituting the morphologic unit enveloped by a single Schwann cell. The presence of abundant lipid droplets in the Schwann cell cytoplasm may have signalled a disturbance of lipid metabolism in these myelinating cells, particularly as lipid comprises about 70% of the myelin in peripheral nerves (cholesterol, galactosphingolipids such as cerebrosides, and phospholipids), with 30% being myelin-specific proteins (including myelin basic protein, myelin-associated glycoprotein, protein O, protein 1 and 2, and peripheral membrane protein 22 and protein). Riboflavin deficiency is also known to impair fatty acid oxidation.15,16,18
There appears to be a selective vulnerability of peripheral nerves in avian riboflavin deficiency to demyelination on the basis that this myelin loss was found in sciatic, brachial, and cervical and lumbar spinal nerves, and to a lesser degree in large and medium-sized intramuscular nerves, but not in ventral and dorsal spinal nerve roots or distal intramuscular and subcutaneous nerves. It has been posited that less susceptible nerves could have greater access to blood-borne vitamins by possessing a more permeable blood-nerve barrier, or their Schwann cells could constitute a distinct subset that responds differently to riboflavin deficiency. Alternatively, the more vulnerable nerves may be located in rapid growth sites, imposing greater metabolic demands on these myelinating cells. 18
In the sciatic and brachial nerves of riboflavin-deficient chickens, novel fibroblastic onion bulbs were found. While onion bulbs are observed in many peripheral neuropathies of diverse etiology, and their formation is recognized as being a non-specific response to repeated demyelination and remyelination, these onion bulbs are generally composed of a concentric wrapping of Schwann cell cytoplasmic processes around the nerve fibre. By contrast, the fibroblastic onion bulbs found in riboflavin-deficient chickens showed a striking extension of long cytoplasmic processes of fibroblasts that enveloped myelinated, demyelinated and remyelinated, but not unmyelinated, nerve fibres and were comprised primarily of fibroblasts. These onion bulbs also arose early in the disease process. Fibroblastic onion bulbs occurred concurrently with tomacula formation, became increasingly prominent as demyelination and remyelination progressed, and involved more than one nerve fibre as a compound structure. This endoneurial fibroblastic proliferation was associated with morphologic evidence of metabolic activation in the form of increase rough endoplasmic reticulum and abundant mitochondria. 17
In the second study, 22 one-day-old broiler chickens were fed a riboflavin-deficient diet (1.65 mg/kg) for 52 days. This deficient diet was also fed to another group of birds for 19 days, the diet then being supplemented by riboflavin at a total concentration of 3.6 mg/kg for 14 days. Schwann cells were enlarged due to accumulated myelin debris and there was segmental demyelination. In birds fed the riboflavin deficient diet only, remyelination was evident from day 25 while, in chickens fed the riboflavin-supplemented diet, myelin restitution was almost complete 14 days after commencement of feeding this ration. In the third study, 23 rapidly growing, meat-type chickens were fed a riboflavin-deficient diet (1.8 mg/kg) from one to 40 days of age. Microscopically, Schwann cells were hypertrophied due to an increased content of cytoplasmic organelles and lipid droplets. Large segments of myelin sheath were found protruding into the Schwann cell cytoplasm, a morphologic change which preceded fragmentation of the myelin sheath and segmental demyelination. Remyelination was prominent at later stages of this study.
Riboflavin deficiency has been shown to cause embryonic mortality in chickens. 24 When Rhode Island Red hens were fed a riboflavin-deficient diet, embryonic mortality increased with the duration of the deficiency, with an initial peak of mortality at 15–21 days, followed by consecutive peaks at 8–14 and 1–7 days, reflecting a gradual depletion of this vitamin in the egg. With White Leghorn hens, embryonic mortality increased between 3 and 16 weeks of the deficiency. During the initial stages of riboflavin deficiency, when embryo mortality is relatively low, birds are able to transport sufficient riboflavin to the egg for the embryo to develop normally for 15–21 days. However, as the deficiency becomes more prolonged, the age of embryonic mortality decreases and, after 14 weeks of the deficiency, there is 100% mortality of 1–7 day-old embryos. When riboflavin was added to the diet, there was a rapid decline in early embryonic mortality. 24
A deficiency of riboflavin also causes a metabolic crisis in a strain of Leghorn chickens that abruptly halts embryonic development and results in sudden embryonic death. 25 Due to a recessive, lethal mutation in a gene encoding a riboflavin-binding protein, these chickens are unable to deposit hepatic-derived riboflavin in their eggs. At mid-gestation, chick embryos are almost totally dependent on catabolism of triglycerides stored in the yolk sac for metabolic energy and, when fatty acid oxidation is severely inhibited in these riboflavin-deficient embryos, they depend on glycolysis for ATP production, severely depleting limited glucose reserves and causing death. At autopsy, riboflavin-deficient chickens have severe fatty livers due to excessive lipid accumulation, marked hypoglycemia, massive cutaneous and visceral hemorrhages due to a blood coagulation defect, and aberrant feather development (termed clubbed down). However, riboflavin administration before incubation of these eggs is able to rescue the embryos and, after hatching, chickens absorb adequate riboflavin levels from their diet and develop normally. 25
Tellurium-induced peripheral neuropathy
The only experimental animal model of peripheral nerve demyelination and remyelination that resembles avian riboflavin deficiency is tellurium neurotoxicity in weanling rats.4,26,27 Tellurium is classified as a developmental neurotoxin. These agents interfere with metabolic processes occurring during development which, in the case of tellurium, is inhibition of squalene epoxidase, an obligate enzyme in the biosynthesis of cholesterol. Weanling rats are particularly susceptible to tellurium intoxication because blockage of the synthesis of cholesterol, the major myelin lipid, coincides with the period of maximal myelin production in peripheral nerves. Moreover, Schwann cells do not upregulate cholesterol synthesis in response to tellurium inhibition and, in fact, downregulate production of specific myelin proteins. Both these reactions tended to exacerbate the deleterious effect of tellurium on peripheral nerves. Remyelination occurs rapidly after cessation of tellurium exposure but, even in the continued presence of tellurium, demyelination diminishes after a few days. There is no axonal degeneration in this model, thus permitting the study of Schwann cell-specific responses during myelin breakdown and subsequent remyelination.4,26,27 While weanling rats are most susceptible to tellurium-induced demyelination, adult rats fed a daily diet of tellurium for 30 days can also show demyelination of peripheral nerves, which then remyelinate. 28 During exposure to tellurium, many Schwann cells hypertrophy and revert to an undifferentiated phenotype, but some die. This change to a precursor cell phenotype seems to be activated by uncoordinated myelin synthesis; it also occurs independently of input from degenerating axons as they remain intact after tellurium exposure.4,26,27
Conclusions
Avian riboflavin deficiency in young, rapidly growing chickens constitutes a useful experimental animal model in which to study the pathology and pathogenesis of peripheral nerve demyelination and remyelination. The principal morphologic features of this model are: (1) it is one of very few animal models to reliably reproduce peripheral nerve demyelination and, when riboflavin levels are restored, rapid remyelination; only a subset of peripheral nerves is selectively vulnerable to this demyelinating neuropathy; (2) paranodal tomacula are found early in the course of demyelination and, rather than being a remyelination phenomenon, may, in fact, initiate myelin loss; (3) since axonal degeneration is minimal in affected peripheral nerves, the model is useful to study primary demyelination; (4) it is the only acquired, primary, demyelinating tomaculous neuropathy described to date in animals; and (5) novel fibroblastic onion bulbs are found in this avian riboflavin deficiency model. Since the peripheral, unlike the central, nervous system exhibits a relatively consistent structure and function in all vertebrates, 3 the findings in this avian model could provide useful insights into the pathogenesis of human demyelinating diseases.
A schematic representation of the pathogenesis of peripheral nerve demyelination produced by riboflavin deficiency in young chickens and tellurium exposure in weanling rats is shown in Figure 2. Pathogenesis of peripheral nerve demyelination produced by riboflavin deficiency in chickens and tellurium in rats.
Footnotes
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.