
Abstract
Methylmercury (MeHg) is a neurotoxin that induces neurotoxicity and cell death in neurons. MeHg increases oligodendrocyte death, glial cell activation, and motor neuron demyelination in the motor cortex and spinal cord. As a result, MeHg plays an important role in developing neurocomplications similar to amyotrophic lateral sclerosis (ALS). Recent research has implicated c-JNK and p38MAPK overactivation in the pathogenesis of ALS. Apigenin (APG) is a flavonoid having anti-inflammatory, antioxidant, and c-JNK/p38MAPK inhibitory activities. The purpose of this study is to determine whether APG possesses neuroprotective effects in MeHg-induced neurotoxicity in adult rats associated with ALS-like neuropathological alterations. In the current study, the neurotoxin MeHg causes an ALS-like phenotype in Wistar rats after 21 days of oral administration at a dose of 5 mg/kg. Prolonged administration of APG (40 and 80 mg/kg) improved neurobehavioral parameters such as learning memory, cognition, motor coordination, and grip strength. This is mainly associated with the downregulation of c-JNK and p38MAPK signaling as well as the restoration of myelin basic protein within the brain. Furthermore, APG inhibited neuronal apoptotic markers (Bax, Bcl-2, and caspase-3), restored neurotransmitter imbalance, decreased inflammatory markers (TNF- and IL-1), and alleviated oxidative damage. As a result, the current study shows that APG has neuroprotective potential as a c-JNK and p38MAPK signaling inhibitor against MeHg-induced neurotoxicity in adult rats. Based on these promising findings, we suggested that APG could be a potential new therapeutic approach over other conventional therapeutic approaches for MeHg-induced neurotoxicity in neurobehavioral, molecular, and neurochemical abnormalities.

Introduction
Amyotrophic lateral sclerosis (ALS) is a motor neuron demyelinating disorder that causes spinal nerve degeneration. It is a subset of disease which primarily affects the brain stem, motor cortex, spinal cord, and corticospinal tract. It even disturbs voluntary movements. 1 Amyotrophic lateral sclerosis is a chronic illness that leads to weakness of top and bottom limbs that develop in myotomal distribution disorder, eventually leading to atrophy of weakened limb muscles.2,3 About 50% of ALS patients have a memory disorder, and 5% have dementia. 4 Motor neuron disorders are low in prevalence and occurrence but have a high mortality rate, causing significant impairment. Amyotrophic lateral sclerosis study reported a median prevalence of 4.48 per 1,00,000 and a standardized incidence of 1.68 per 1,00,000 people per year, varying from one geography, sex, and age to another. 5 Searching for therapies that could potentially change the pathogenic mechanism associated with ALS could improve ALS-mediated motor neuron dysfunction. 6
The disease ALS is linked to motor neuron degeneration, 7 impaired axonal transmission, 8 and dysregulated processes, including aberrant ribonucleic acid metabolism. 9 Glutamate excitotoxicity, 10 impaired protein homeostasis, 11 oxidative stress, 12 and mitochondrial dysfunction 13 are additional causal factors in the progression of ALS. In addition, the proliferation of astroglial and microglial cells eventually contributes to neuronal degeneration and upregulation of mitogen-activated protein kinase (MAPK), increased oxidative deoxyribonucleic acid (DNA) damage to motor neurons. 14
Neurotoxins such as methylmercury (MeHg), 15 b-N-Methylamino-L-Alanine (L-BMAA), 16 β-Sitosterol-β-d-glucoside (BSSG), 17 Bisphenol A (BPA), 18 and superoxide dismutase-1 (SOD1) transgenic models, 19 TDP43-Q331K 20 and FVB-C9orf, are well-established validated experimental animal models for ALS assessment.
The current study utilizes MeHg, a potent neurotoxin, easily penetrating the neural tissue and mimicking ALS-like behavioral and neurochemical alterations.15,21 MeHg’s neuropathogenic mechanism directly damages the central nervous system glial cells called astrocytes, resulting in neuroexcitation, motor neuron degeneration,22, and demyelination. 23 The formation of free reactive nitrogen species and reactive oxygen species-mediated radicals leads to changes in DNA sequences, increased lipid peroxidation, glutamate excitotoxicity, and calcium homeostasis. 24 These disease factors can result in cell death by different intracellular signaling networks such as the c-JNK/P38MAPK pathway. 25
Mitogen-activated protein kinases are a group of related serine/threonine kinases that integrate diverse signals that guide cell response to proliferative or stressful stimuli. 26 The MAPK family includes c-JNK or stress-activated protein kinase (JNK/SAPK), p38MAPK, and extracellular signal-regulated kinase. 27 Activation of c-JNK is often associated with neuronal degeneration. It has recently been demonstrated that it occurs in neurodegenerative illnesses such as ALS, Parkinson’s disease (PD), Alzheimer’s disease (AD), and Huntington’s disease (HD).28-35 Some studies have documented that inhibition of c-JNK attenuates motor neuron death 28 and protects dopaminergic neurons in PD. 36 It also improves neurological dysfunction, decreases infarctions in cerebral ischemia, 37 and improves neuroprotection in AD. 38
P38MAPK includes different factors, such as cytoskeleton protein phosphorylation and cytokine biosynthesis. Nitric oxide pathway is believed to play a function in motor neuron degeneration and axonal transport defects in ALS.39,40 P38MAPK inhibition has also been shown to be effective in amyotrophic lateral sclerosis41-43 and various neurodegenerative diseases, for example AD, PD, peripheral neuropathy, multiple sclerosis (MS), and epilepsy.44-49 Besides, increase in p38MAPK is a major neurocomplication risk factor for ALS.39,50,51
Apigenin (APG) is a flavonoid found in many vegetables and herbal varieties such as parsley, celery, basil, chamomile, cilantro, and oregano. 52 It already has a broad spectrum of biological functions such as anticancer, antiviral, antibacterial, antioxidant, neuroprotective, and strong anti-inflammatory action.53-58 Apigenin has also been confirmed to take part in neuroprotective function in various neurological disorders, such as PD, AD, and dementia.57-65
Besides this, APG also exhibits neuroprotection against cerebral ischemia, subarachnoid hemorrhage, MS, epilepsy, and depression.66-78
Apigenin has also been identified as an inhibitor of c-JNK/p38MAPK in several diseases 79 , including AD,58,59,80 PD,60,81 diabetic neuropathy, 82 ischemia/reperfusion, 83 and renal injury. 84 The C-JNK/p38MAPK inhibition has been investigated to treat a variety of central nervous system diseases due to its efficient lifespan and distinct function. Therefore, the current research’s main objective was to study apigenin’s neuroprotective properties on neurobehavioral, biochemical, and gross pathological abnormality in methylmercury-treated ALS-like neuropathological abnormalities in adult rats and, in particular, to demonstrate the upregulation of the c-JNK and p38MAPK signaling pathways in the pathogenesis of ALS.
Materials and methods
Experimental animals
A total of 36 rats were used in the current study. All experiments were performed on six-month-old adult Wistar rats (weight 180–200 g) obtained from the Central Animal House, ISF College of Pharmacy in Moga, Punjab, India. Rats were housed under acclimatized laboratory conditions in polyacrylic cages (23°C ± 2° C rooms and 60 ± 10% RH) in controlled air with proper food and water ad libitum facilities. From 9:00 a.m. to 5:00 p.m., all behavioral parameters were evaluated. The Project was approved as number 816/PO/ReBiBt/S/04/CPCSEA as IAEC/CPCSEA/M 25/2019/Protocol No 423, by the Institute for Animal Ethics Committee (IAEC), in accordance with the guidelines provided by the Indian government.
Drugs and chemicals
Sigma-Aldrich supplied the MeHg (USA). APG (protocol drug) was provided as an ex-gratia sample by BAPEX, New Delhi, India. The chemicals utilized for the evaluation of neurotransmitters are of laboratory grade, and other biochemical markers (analytical grade) were procured. Chemicals purchased from Himedia, Mumbai, India, are acetylcholine, EDTA, sodium-di-hydrogen phosphate, sulfanilamide, sulfosalisylic acid, tri-chloroacetic acid, Tris-HCL, Tris-sodium citrate dehydrate, CMC, 1-heptane sulfonic acid, and perchloric acid. Chemicals purchased from SDFCL, Haryana, India, are di-chromatic acetic acid, di-nitrophenyl hydrazine, ethyl acetate, formaldehyde, liquid ammonia, potassium-di-hydrogen phosphate, acetonitrile, and methanol. Chemicals purchased from CDH, New Delhi, India, are sodium acetate, disodium hydrogen phosphate, sodium hydroxide, sodium succinate, succinic acid, hydrogen peroxide, xylene, Tris-buffer, disodium hydrogen O-phosphate, and di-potassium hydrogen phosphate. Chemicals purchased from Sigma, Missouri, USA, are dithiobis-nitrobenzoic acid (DTNB) and sodium dodecyl sulfate. Chemicals purchased from Loba, Mumbai, India, are N-ethylenediamine dihydrochloride, sodium nitrite, and thiobarbituric acid. Before usage, the drug and chemicals were carefully prepared. The purity of APG is > 95.0% (HPLC). Apigenin was dissolved in water with 1% CMC and administered orally (p.o.). 85
Experimental protocol schedule
The experimental protocol schedule was of 42 days. MeHg was administered orally from day 1 to day 21. APG was administered daily from day 22 to day 42. The MeHg dose was taken from prior studies.
15
There were no standard drugs used in the present study. Rats were equally distributed into six groups (each group contained n = 6): group 1–normal control; group 2–vehicle control; group 3–APG perse (80 mg/kg, p.o.); group 4–MeHg (5 mg/kg, p.o.); group 5–MeHg (5 mg/kg, p.o.) + APG (40 mg/kg, p.o.); and group 6–MeHg (5 mg/kg, p.o.) + APG (80 mg/kg, p.o). Different behavioral evaluations were conducted between days 1 and 42. All rats were sacrificed by decapitation on day 43. All the isolated brains were used for analysis of neurochemical, neuroinflammatory, and neurotransmitters. The experimental protocol schedule is summarized in Figure 1. Experimental protocol schedule.
Experimental animal model of MeHg-induced neurotoxicity in adult rats
The MeHg-induced preclinical animal model of ALS in adult rats was developed as per the standardized method established by Minj et al. (2021) 3 and Alam et al. (2020). 15 MeHg was regularly administered to adult Wistar Rats by oral gavages continued to 21 days at 5 mg/kg of dose. MeHg is a potent neurotoxin capable of quickly penetrating the neural tissue and was similar to ALS. 86 In this study, we utilized MeHg for observing behavioral and neurochemical alterations in rats.
Parameters
Measurement of body weight
The body weight was measured on the 1st, 7th, 14th, 21nd, 28th, 35th, and 42nd days of the experiment schedule. 87
Behavioral parameters
Morris water maze task
The Morris water maze test has been carried out to evaluate memory and cognitive impairment. The escape latency (ELT) was recorded by utilizing MWM on the protocol schedule’s 39th, 40th, 41th, and 42nd days.88,89 Time taken by rats to reach the target platform is known as ELT, which is indicated in seconds. On the 42nd day, rats were permitted to swim independently in a pool containing a hidden platform, for 120 s, and time spent in the target quadrant (TSTQ) was recorded. The TSTQ reflects the memory consolidation after learning.
Grip strength test
Grip strength enables rats to perform neuromuscular functions by measuring grip strength through the maximum picking force generated by the forelimb; rats are kept on the tail and reduced toward the unit Kg until a handle with both fronts is grabbed. The rats have been drawn slowly back till they have been gripped by the handle. The grip strength was measured out at different time intervals during 1st, 22nd, 32nd, and 42nd days of the experimental period. Three consecutive trials were held for each session. The force needed to remove the rat from the handle was automatically measured by the equipment. 89
Forced swim test
The forced swim test is well known as an antidepressant activity for rodent evaluation. The immobility time was recorded after treatment on the 21st, 28th, 35th, and 42nd days. Each animal was tested for 5 min in the cylinder. During the time, only small moves were made by the rat to keep its heads above the water, and the moves, latency to float, and floating duration were recorded. Rats were forced to swim in an unavoidable condition during the forced swim procedure. The animal becomes immobile or only makes the necessary movement to keep his head above water following an intense period of struggle. In this test, the immobility observed reflects a state of despair. 90
Open field test
An open field apparatus was utilized to assay the experimental locomotor activity and anxiety-mimicking behavior. After the equipment was prepared, a single rat was placed into the center of the area; time was started immediately, and exploratory behavior was permitted for 5 min. The silence was maintained during the testing period. The rat was returned to its cage once the test was completed. The number of crossing lines by rats (number of lines crossed by four paws) and rearing was monitored for 5 minutes, and the locomotive activity indicator was adopted. The toxin test was conducted five times, and treatment days were 1st, 11th, 21st, 31st, and 41st days. 91
Neurochemical parameters
Preparation of brain homogenate
All rats were sacrificed, and fresh brains were isolated on the 43rd day of the protocol schedule. The isolated brains were cleaned by ice-cold isotonic saline solution, and the homogenate was made using (w/v) 0.1 M phosphate buffer (pH 7.4). The homogenate solution was acquired by centrifuging 15 min with 10,000 g, supernatant, and aliquots used to estimate biochemicals. 92
Evaluation of cellular and biochemical marker
c-JNK levels
The levels of c-JNK were estimated in the brain homogenate using ELISA testing kits Elabsciences, Wuhan, Hubei, China. By following standard procedure, the test was carried out. The values were calculated as pg/mL protein. 93
p38MAPK levels
The p38MAPK levels in rat brain homogenate were determined by using ELISA kits Elabsciences, Wuhan, Hubei, China. By using the standard procedure, ELISA tests were performed. The values were measured as pg/mL of protein. 94
Myelin basic protein levels
The MBP was estimated by ELISA testing kits. As per the given standard procedure, the test was carried out using sample homogenate (E-EL-R0642/MBP; Elabsciences, Wuhan, Hubei, China). The units were indicated as μg/mg protein. 95
Assessment of apoptotic markers
Caspase-3, Bax, and Bcl-2 levels
The brain homogenate was utilized to evaluate caspase-3 levels. 96 According to the standard procedure in ELISA testing, the Bax 97 and Bcl-2 98 levels were determined (E-EL-R0160/caspase-3; E-EL-R0098/Bax/Bcl2Elabsciences, Wuhan, Hubei, China).
Neurotransmitter’s evaluation
Gamma-aminobutyric acid and glutamate levels
The quantitative analysis was conducted on the tissue samples using the Jamal and collaborating process. Glutamates and gamma-aminobutyric acid (GABA) were quantified after derivation using o-phthalaldehyde/β-mercaptoethanol. The supernatant neurotransmitter was shown to be ng/mg protein. 99
Acetylcholine levels
Acetylcholine was estimated using an ELISA kit (KRISHGEN Diagnostics, India). The kit’s instructions were followed when preparing all the samples and reagents. The absorbance of the reactive mixture was measured at 540 nm by using the microtiter plate. The supernatant’s neurotransmitter was indicated in ng/mg protein. 100
Serotonin levels
The brain homogenate was utilized to measure the serotonin level by using high-performance liquid chromatography (HPLC). This moving phase includes acetonitrile sodium citrate buffer (pH4.5). The buffer contains 10 Mmol/I citric acid, 25 Mmol/l of NaH2HPO4, 25 Mmol/l of EDTA, and two Mmol/l of sulfonic acid one heptane. Experimental electrochemical situations ranged from 5 nA to 50 nA and were + 0.75 V. The separation process has been performed at a 0.8 mL/min flow rate. The manual injection was made of 20 μL of samples. On experiment day, brain samples were standardized with 0.2 mol/l of perchloric acid. Afterward, centrifugation was started for 5 minutes at 12,000 g. The standard curve of serotonin concentrations was calculated using 10–100 mg/mL standard solution. 101
Measurement of neuroinflammatory markers
TNF-α and IL-1β levels
An immunoassay rat’s kit has been used to determine the level of TNF-α 102 and IL-1β 103 by using an immune testing kit (E-EL-R0019/TNF-α; E-EL-R0012/IL-1β; ELabSciences, Wuhan, Hubei, China). TNF-α and IL-1β are expressed as pg/mg protein.
Measurement of oxidative stress marker levels
Acetylcholinesterase levels
The method described was used to measure AchE activity quantitatively within the brain. 104 The test mix contained a supernatant content of 0.15 mL, a buffer of 0.01 M of sodium phosphate (pH = 8), a thiocholine iodide of acetyl 0.10, and a DTNB of 0.10 mL (Ellman reagent). At a wavelength of 412 nm, changes in spectrophotometric absorption were immediately measured. The enzymes were expressed as mM/mg protein for supernatant activity.
Reduced glutathione levels
The method described was used to estimate reduced glutathione in the brain. 105 One mL of supernatant with 1 mL of sulfosalicylic acid at cold temperature of about 4°C had been precipitated for an hour. At 1200 Tog for 15 min, the samples are centrifuged. In addition, 2.7 μL (0.1 M, pH 8) of DTNB was added (0.2 μL) in 1 mL of supernatant. The developed yellow color was measured with a spectrophotometer immediately at 412 nm. Glutathione is measured as a U/mg protein.
Thiobarbituric acid reactive substances
According to Mehan et al., 2020, thiobarbituric acid reactive substances (TBARS), a lipid peroxidation end product, are measured quantitatively in homogenous brain tissue. The quantity of TBARS was assessed by using the spectrophotometer at 532 nm following its reaction with thiobarbituric acid. U/mg protein is the unit for value expressed in TBARS levels. 106
Superoxide dismutase levels
The UV/VIS spectrophotometer has been tested for superoxide dismutase (SOD) activity. The testing system includes 50 mml/L sodium carbonate, 0.1 mmol/L EDTA, 96 mmol/L tetrazolium-nitro blue, and 2 mL (0.05 mL) of hydroxylamine taken in the cuvette and measured at 560 nm by using the auto-oxidative method (PERKIN ELMER UV/VIS Spectrophotometer) for 2 min with 30 s interval. SOD activity is shown in U/mg protein. 107
Nitrite levels
Griess reagent was used to estimate nitrite levels (0.1% N-ethylenediamine dihydrochloride, 2.5% phosphoric acid, and 1% sulfanilamide); in a colorimetric test, nitrite accumulation in the supernatant is determined as an indicator of nitrate oxide. Samples were incubated for 10 min in the dark closed area at room temperature and absorbent in 540 nm wavelength—the equivalent volumes of Griess reagent and supernatant were mixed. A standard sodium nitrite curve assesses the supernatant’s nitrite level as mM/mg protein. 108
Lactate dehydrogenase levels
To estimate the lactate dehydrogenase (LDH) level, the spectrophotometric analysis method was utilized. Lactate dehydrogenase catalyzes lactate oxidation and reduces nicotinamide adenine dinucleotide (NAD) to NADH at the same time. Serum LDH activity is equivalent to a rise in absorption when NAD reduces. A diagnostic kit for lactate dehydrogenase activities was employed (Coral Diagnostics, Indian) in rat’s homogenous brains and in U/mg. 109
Total protein estimation
The Coral protein kit was utilized to measure the volume of total protein in the sample (biuret method). 110
Gross pathological examination of rat brains
On the 43rd day, the decapitation method was used to sacrifice the rats; brains were isolated for gross pathological analysis. The entire rat brain was used to take the coronal sections. On glass slides, 2-mm thick brain sections (coronally to the back of the cerebral cortex from the anterior pole) were placed. 111 A digital camera was used to visualize all the brain regions. Each brain segment’s demyelination region (mm) was evaluated on the 43rd day of the experiment using MOTICAM-BA310 image plus 2.0 analysis software. 112 The demyelination volume (mm3) was measured by converting the demyelination region for each segment of the coronal brain (mm).113,114 Each brain section on the 43rd-day image study was taken to detect the demyelination size (mm3) in the dark gray area near the striatum. The injury size was estimated by measuring the demyelination area of each 2-mm thick coronal (l × b × h) brain section.115,116
Statistical analysis
The post hoc Bonferroni test was used for two-way ANOVA, and similarly, post hoc Tukey’s multi comparison test was used for one-way ANOVA. The significant data were analyzed by considering p < 0.01. To check the normal distribution of data and estimate sample size, the Kolmogorov Smirnov test was used. GraphPad Prism version 5.03 for Windows (GraphPad Software, San Diego, CA, USA) was used to generate all statistical results. The statistical values were expressed as mean ± standard error of mean (SEM).
Results
Neuroprotective effect of apigenin on body weight in MeHg-induced neurotoxicity in adult rats
On 1st, 7th, 14th, 21st, 28th, 35th, and 42nd days, the body weight was measured to observe the protective effect of the APG in MeHg-treated adult rats. On day 1, no considerable changes were found between the treatment groups. While comparing to the normal, vehicle, and APG 80 mg/kg perse groups, the body weight was dramatically reduced in MeHg-treated rats. In contrast with the MeHg-treated group, reduced body weight was successfully recovered on 35th and 42nd days of the post-treatment with an ongoing dose of APG 40 mg/kg and APG 80 mg/kg [two-way ANOVA: F (30,180) = 304.5, p < .01]. An efficient restoration in body weight was observed in rats administered by APG 80 mg/kg (Figure 2). Neuroprotective effect of apigenin on the restoration of body weight in MeHg-induced neurotoxicity in adult rats. APG treatment restored the body weight and expressed in grams. Statistical analysis followed by two-way ANOVA (post hoc Bonferroni’s test). Values expressed as mean ± SEM (n = 6 rats per group). * p < .01 v/s normal control, vehicle control, and APG 80 perse; @ p < .01 v/s MeHg; @# p < .01 v/s MeHg + APG 40.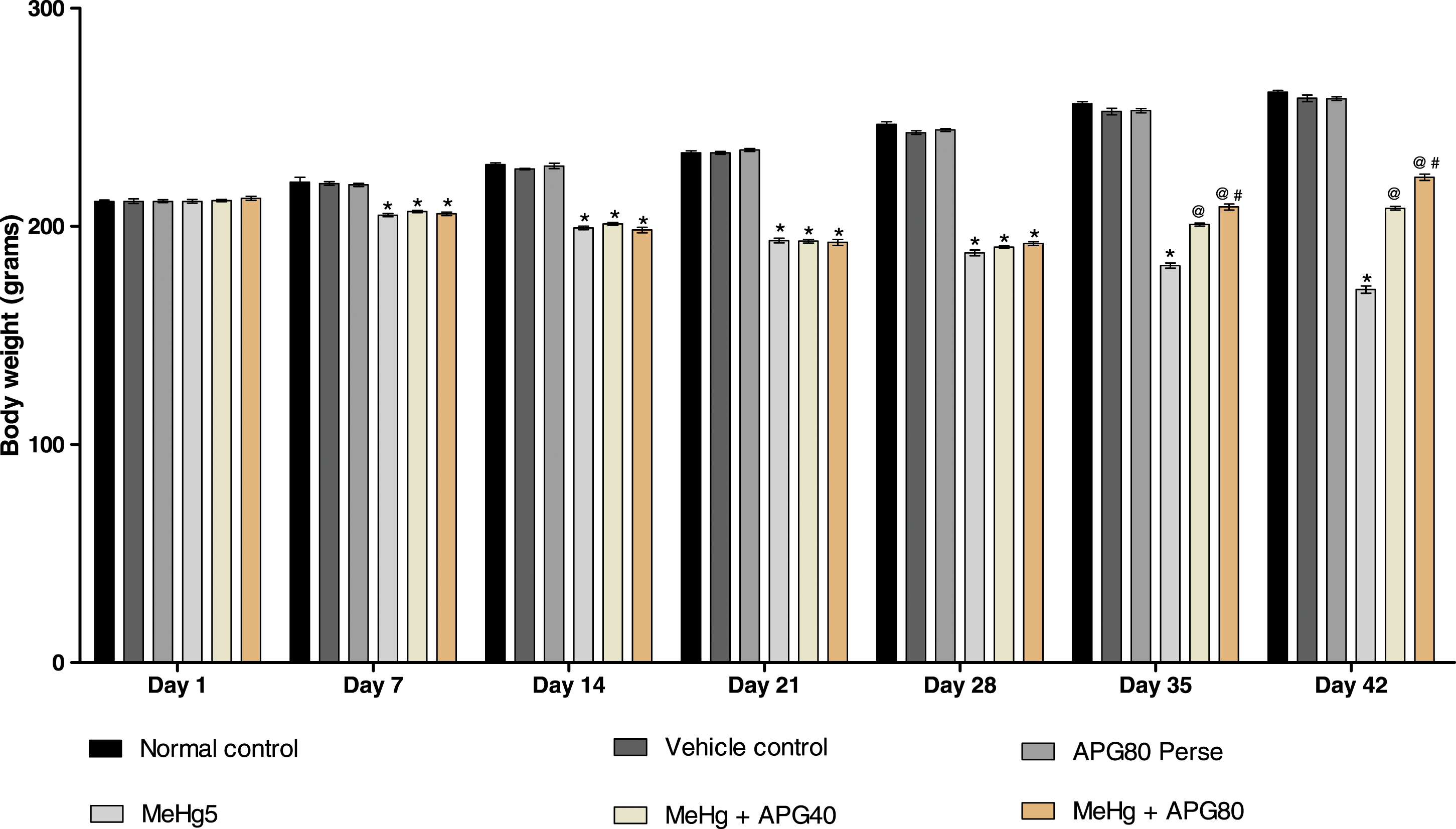
Neuroprotective effect of apigenin in the prevention of behavioral abnormalities in MeHg-induced neurotoxicity in adult rats
Improved memory and cognitive function after prolonged treatment with apigenin
The escape latency was assessed on 38th, 39th, 40th, and 41st days of the experimental schedule. According to the findings, continuous treatment of MeHg-treated rats progressively enhances ELT compared to the normal group. APG 80 mg/kg perse-administered rats show no notable changes in escape latency as compared with normal and vehicle control groups. In contrast to MeHg-administered group, long-term administration of APG 40 mg/kg and APG 80 mg/kg for 21 days shows dose-dependent reduction in ELT [two-way ANOVA: F (10,60) = 60.93, p < 0.01]. In addition, APG 80 mg/kg treatment rats restored long-term memory more effectively than 40 mg/kg APG-administered rats (Figure 3). Neuroprotective effect of apigenin on escape latency time using Morris water maze in MeHg-induced neurotoxicity in adult rats. APG treatment restored the escape latency time and expressed in seconds. Statistical analysis followed by two-way ANOVA (post hoc Bonferroni’s test). Values expressed as mean ± SEM (n = 6 rats per group). * p < .01 v/s normal control, vehicle control, and APG 80 perse; @ p < .01 v/s MeHg; @# p < .01 v/s MeHg + APG 40.
On 42nd day, the time spent in the TSTQ was performed in all groups. The MeHg-exposed rats gradually decreased TSTQ compared to the normal, vehicle, and APG 80 mg/kg perse treatment groups. In contrast, prolonged treatment with APG 40 and APG 80 mg/kg considerably increased TSTQ compared to the MeHg-induced ALS-like symptoms in adult rats [one-way ANOVA: F (5,25) = 1.264, p < .01]. Effective long-term memory restoration was found in the APG 80 mg/kg treatment group (Figure 4). Neuroprotective effect of apigenin on TSTQ using Morris water maze in MeHg-induced neurotoxicity in adult rats. APG treatment increases cognitive functions by restoring TSTQ and expressed in seconds. Statistical analysis followed by one-way ANOVA (post hoc Tukey’s test). Values expressed as mean ± SEM (n = 6 rats per group). * p < .01 v/s normal control, vehicle control, and APG 80 perse; @ p < .01 v/s MeHg; @# p < .01 v/s MeHg + APG 40.
Improved locomotion abnormalities after prolonged treatment with apigenin
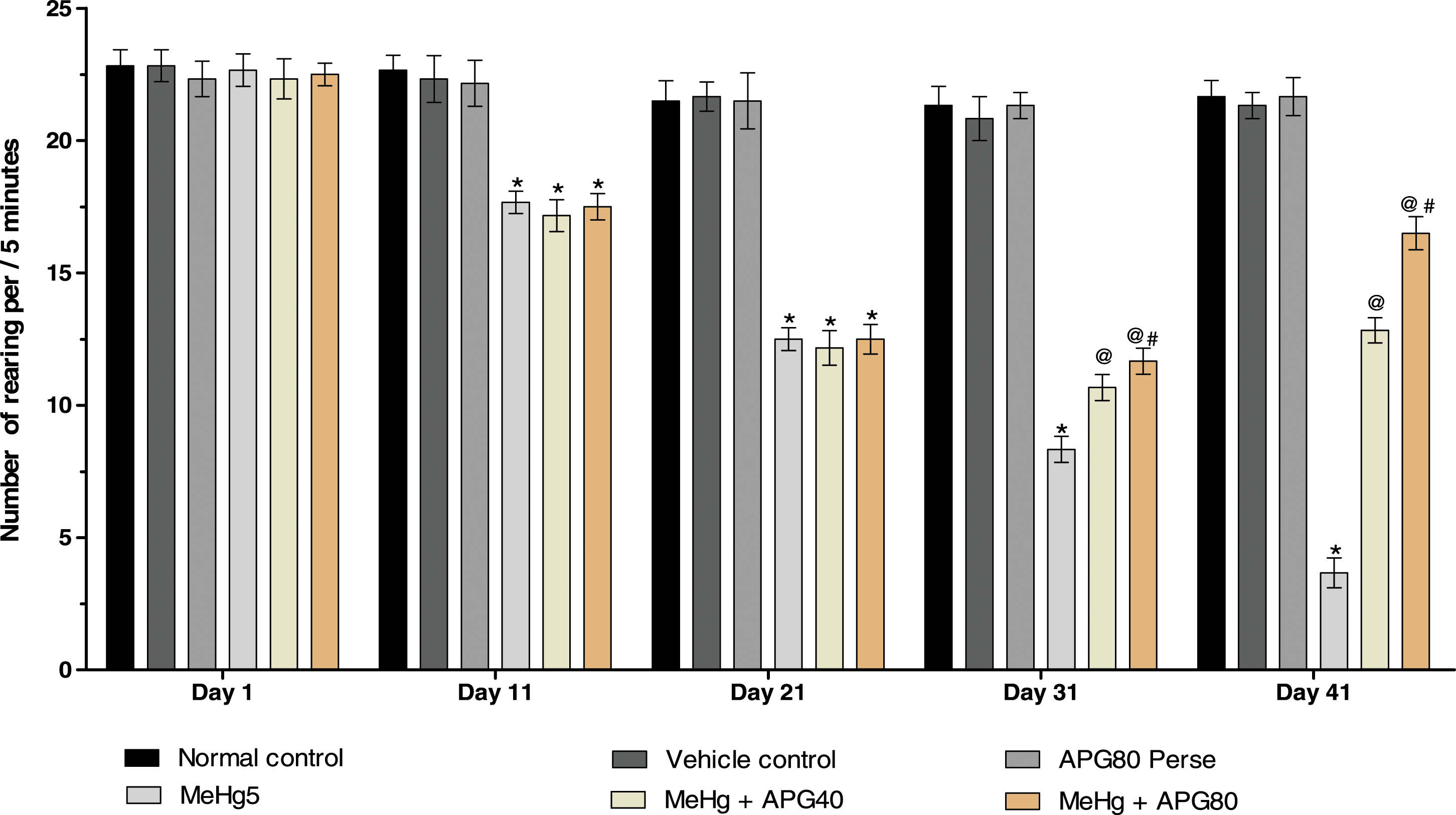
The locomotor activity was measured in rats using open field apparatus on the 22nd, 28th, 34th, and 40th days of the protocol schedule. The MeHg-treated group on the 21st day showed a significant decrease in locomotor activity compared to the normal, vehicle, and APG 80 mg/kg treatment groups. Chronic administration with APG 40 mg/kg- and APG 80 mg/kg-treated groups shows a considerable improvement in the locomotive activity and the number of rearing activities as compared with MeHg-exposed rats [two-way ANOVA: F (20,120) = 31.43, p < .01]. The high-dose APG 80 mg/kg-treated rats more effectively improved the locomotion and rearing activity as compared to APG 40 mg/kg-treated rats [two-way ANOVA: F (20,120) = 23.18, p < .01] (Figures 5 and 6). Neuroprotective effect of apigenin on locomotion activity in MeHg-induced neurotoxicity in adult rats. APG treatment increased the locomotor activity and expresses the ratio of the number of boxes crossed per 5 min. Statistical analysis followed by two-way ANOVA (post hoc Bonferroni’s test). Values expressed as mean ± SEM (n = 6 rats per group). *p < .01 v/s normal control, vehicle control, and APG 80 perse; @ p < .01 v/s MeHg; @# p < .01 v/s MeHg + APG 40. Neuroprotective effect of apigenin on locomotion (number of rearing) in MeHg-induced neurotoxicity in adult rats. APG treatment increase locomotion by restoring the number of rearing and expressed as the ratio of the number of boxes crossed per 5 min. Statistical analysis followed by two-way ANOVA (post hoc Bonferroni’s test). Values expressed as mean ± SEM(n = 6 rats per group). *p < .01 v/s normal control, vehicle control, and APG 80 perse; @ p < .01 v/s MeHg; @# p < .01 v/s MeHg + APG 40.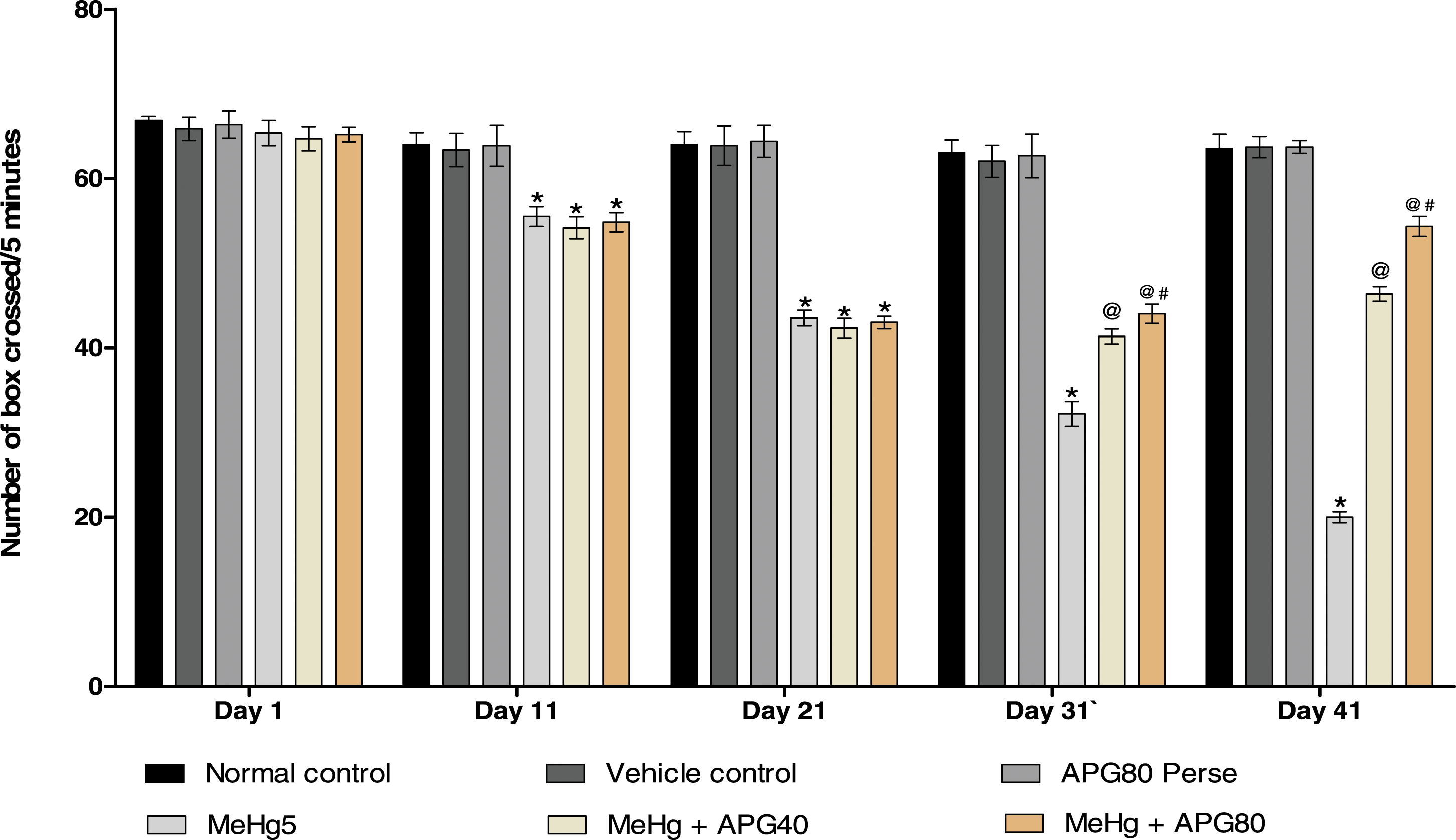
Improved grip strength after prolonged treatment with apigenin
The grip strength test was conducted on 1st, 22nd, 31st, and 41st days of the experimental protocol schedule. On the 1st day, no notable changes were found among all groups. MeHg-treated rats demonstrate significantly lower grip strength force compared to the normal, vehicle, and APG 80 mg/kg perse groups. On the day 22, Prolonged administration with APG 40 and 80 mg/kg for 21 days significantly increased grip strength force comparison to the MeHg-induced ALS-like symptoms in adult rats on the 32nd and 42nd days and restores grip strength force [two-way ANOVA: F (15,90) = 322.9, p < .01]. Moreover, APG 80 mg/kg-administrated rats effectively improved gripping than the APG 40 mg/kg-treated rats (Figure 7). Neuroprotective effect of apigenin on grip strength test in MeHg-induced neurotoxicity in adult rats. APG treatment increases the grip strength force by restoring the grip strength and expressed in kgf. Statistical analysis followed by two-way ANOVA (post hoc Bonferroni’s test). Values expressed as mean ± SEM (n = 6 rats per group). *p < .01 v/s normal control, vehicle control, and APG 80 perse; @ p < .01 v/s MeHg; @# p < .01 v/s MeHg + APG 40.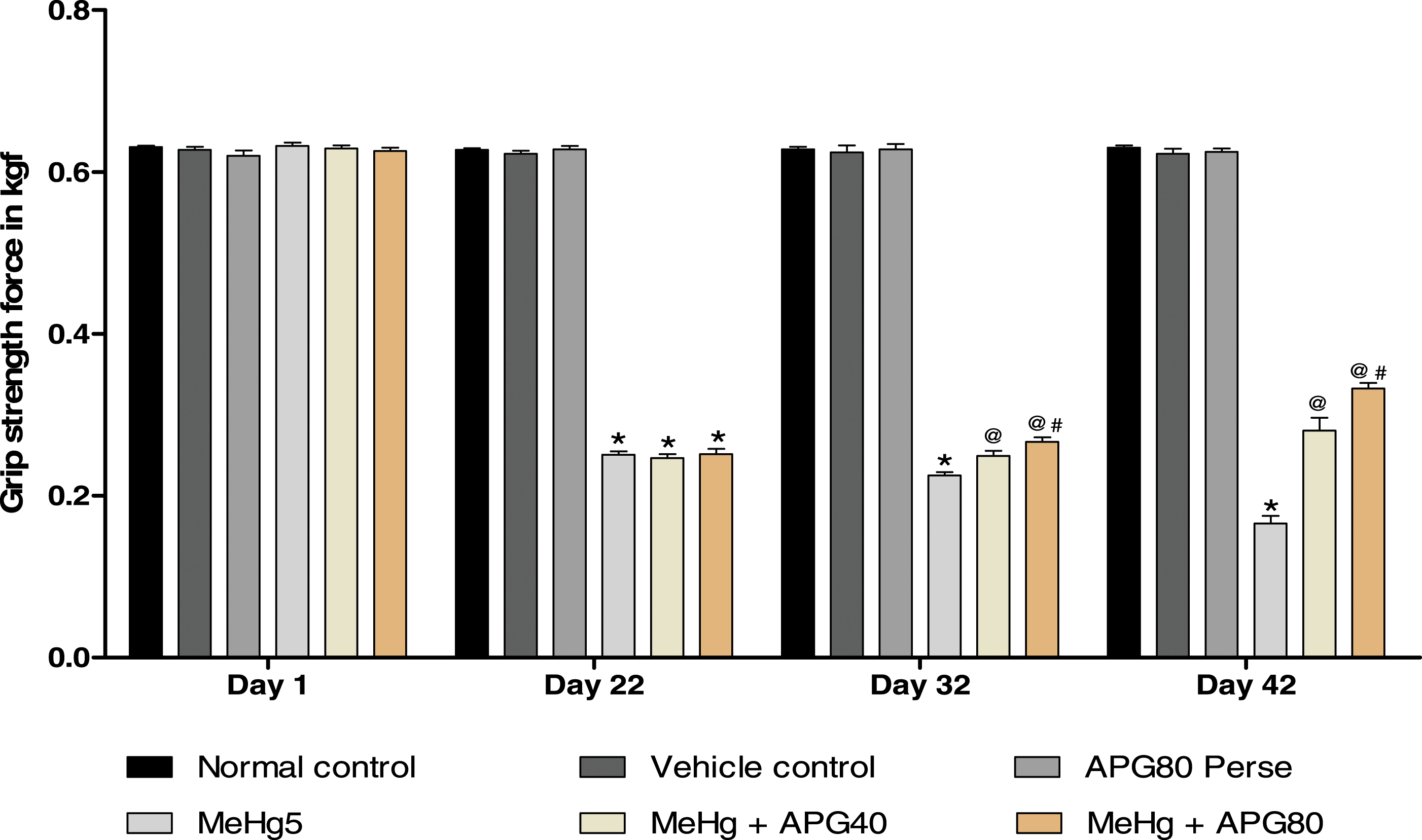
Reduced depression-like activity after prolonged treatment with apigenin
The forced swim test was conducted to assess the immobility activity of rats on 21st, 28th, 35th, and 42nd days of the experiment. On day 1, no substantial changes were noted between all groups. The MeHg-exposed rats showed increased immobility time and depression-like activity in the forced swim test. The MeHg-treated group showed more mobility than the normal, vehicle, and APG 80 perse groups. Chronic administration of APG 40 mg/kg- and 80 mg/kg-administered group resulted in a remarkable reduction in the immobility time in comparison to the normal, vehicle, and APG 80 perse treatment groups [two-way ANOVA: F (15,90) = 21.66, p < .01] (Figure 8). Neuroprotective effect of apigenin on immobility in MeHg-induced neurotoxicity in adult rats. APG treatment decreases depression-like activity by restoring immobility and expressed in seconds. Statistical analysis followed by two-way ANOVA (post hoc Bonferroni’s test). Values expressed as mean ± SEM (n = 6 rats per group). *p < .01 v/s normal control, vehicle control, and APG 80 perse; @ p < .01 v/s MeHg; @# p < .01 v/s MeHg + APG 40.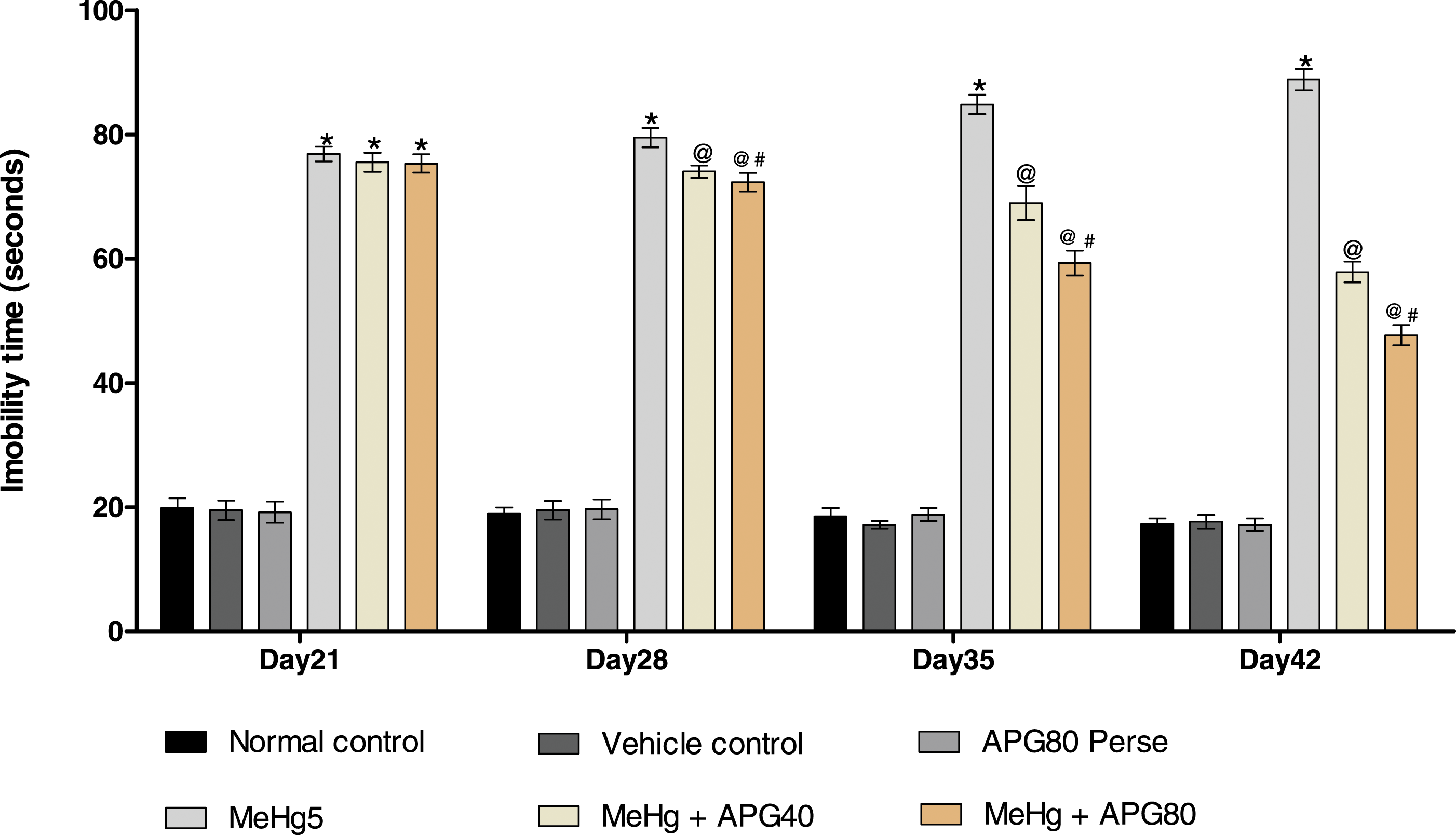
Neuroprotective effect of apigenin on neurochemical alterations in MeHg-induced neurotoxicity in adult rats
Decreased level of c-JNK after prolonged treatment with apigenin
Neuroprotective effect of APG on the c-JNK, p38MAPK, and MBP levels in MeHg-induced neurotoxicity in adult rats.

Statistical analysis followed by one-way ANOVA (post hoc Tukey’s test). Values expressed as mean ± SEM (n = 6 rats per group). *p < .01 v/s normal control, vehicle control, and APG 80 perse; @ p < 0.01 v/s MeHg; @# p < .01 v/s MeHg + APG 40.
Decreased level of p38MAPK after prolonged treatment with apigenin
The p38MAPK level was measured in the rat brain homogenate on the 42nd day of the experiment. Chronic MeHg treatment led to a substantial rise in p38MAPK levels compared to the normal, vehicle, and APG 80 mg/kg perse groups. The level of p38MAPK protein decreased significantly after treatment with APG 40 mg/kg and APG 80 mg/kg than the MeHg exposure rats [one-way ANOVA: F (5,25) = 0.759, p < .01] (Table 1(b)).
Restoration of myelin basic protein level after prolonged treatment with apigenin
The brain homogenate was used to determine the MBP level by utilizing an ELISA kit. Chronic administration of MeHg showed significant MBP level reductions compared to the normal, vehicle, and APG 80 perse groups. In conclusion, the long-term administration of APG 40 mg/kg and APG 80 mg/kg in rats led to a substantial restoration of MBP level than the MeHg-treated rats [one-way ANOVA: F (5,25) = 0.317, p < .01]. The remarkable restoration of myelin basic protein was observed when treating the rats with 80 mg/kg APG (Table 1(c)).
Restoration of caspase-3, Bax, and Bcl-2 levels after prolonged treatment with apigenin
Neuroprotective effect of apigenin on apoptotic markers in MeHg-induced neurotoxicity in adult rats.

Statistical analysis followed by one-way ANOVA (post hoc Tukey’s test). Values expressed as mean ± SEM (n = 6 rats per group). *p < .01 v/s normal control, vehicle control, and APG 80 perse; @ p < .01 v/s MeHg; @# p < .01 v/s MeHg + APG 40.
Restoration of neurotransmitter level after prolonged treatment with apigenin
Neuroprotective effect of apigenin on neurotransmitter level in MeHg-induced neurotoxicity in adult rats.
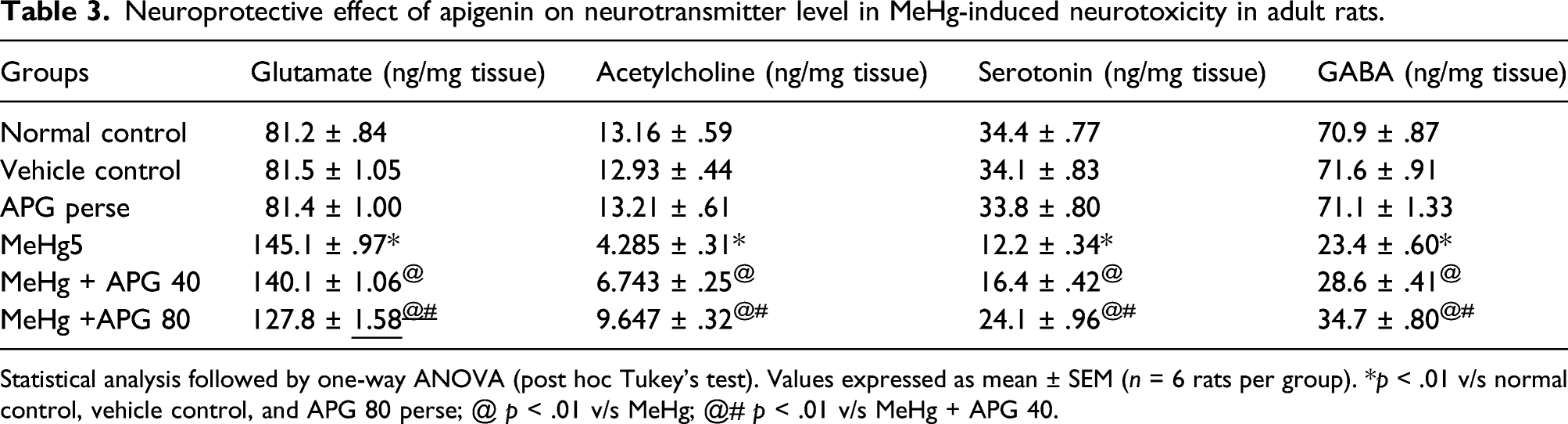
Statistical analysis followed by one-way ANOVA (post hoc Tukey’s test). Values expressed as mean ± SEM (n = 6 rats per group). *p < .01 v/s normal control, vehicle control, and APG 80 perse; @ p < .01 v/s MeHg; @# p < .01 v/s MeHg + APG 40.
Decreased neuroinflammatory cytokines after prolonged treatment with apigenin
Neuroprotective effect of apigenin on neuroinflammatory cytokines in MeHg-induced neurotoxicity in adult rats.

Statistical analysis followed by one-way ANOVA (post hoc Tukey’s test). Values expressed as mean ± SEM (n = 6 rats per group). *p < .01 v/s normal control, vehicle control, and APG 80 perse; @ p < .01 v/s MeHg; @# p < .01 v/s MeHg + APG 40.
Restored antioxidant levels after prolonged treatment with apigenin
Neuroprotective effect of apigenin on oxidative stress markers in MeHg-induced neurotoxicity in adult rats.

Statistical analysis followed by one-way ANOVA (post hoc Tukey’s test). Values expressed as mean ± SEM (n = 6 rats per group). *p < .01 v/s normal control, vehicle control, and APG 80 perse; @ p < .01 v/s MeHg; @# p < .01 v/s MeHg + APG 40.
Neuroprotective effect of apigenin on gross pathological alterations and demyelination in MeHg-induced neurotoxicity in adult rats
Improved whole-brain morphological alterations after prolonged treatment with apigenin
The rat brains treated with MeHg had tissue damage, loss of myelin sheath, degeneration of motor neurons and spinal cord, including significant axon loss, swelling, demyelination, and meninges breakage compared to the normal, vehicle, and APG perse groups. The normal, vehicle, and APG (80 mg/kg)-treated groups displayed an optimally regular shape, undamaged form with clearly visible meninges. In MeHg-induced rats, continuous administration with APG 40 mg/kg restored morphological alterations. The high dose of APG 80 mg/kg-treated rat brain represents significant restoration in demyelination (Figure 9). Neuroprotective effect of apigenin in the improvement of whole-brain morphological alterations in MeHg-induced neurotoxicity in adult rats: (a) normal control; (b) vehicle control; (c) APG perse; (d) MeHg5; (e) MeHg + APG 40; and (f) MeHg + APG 80.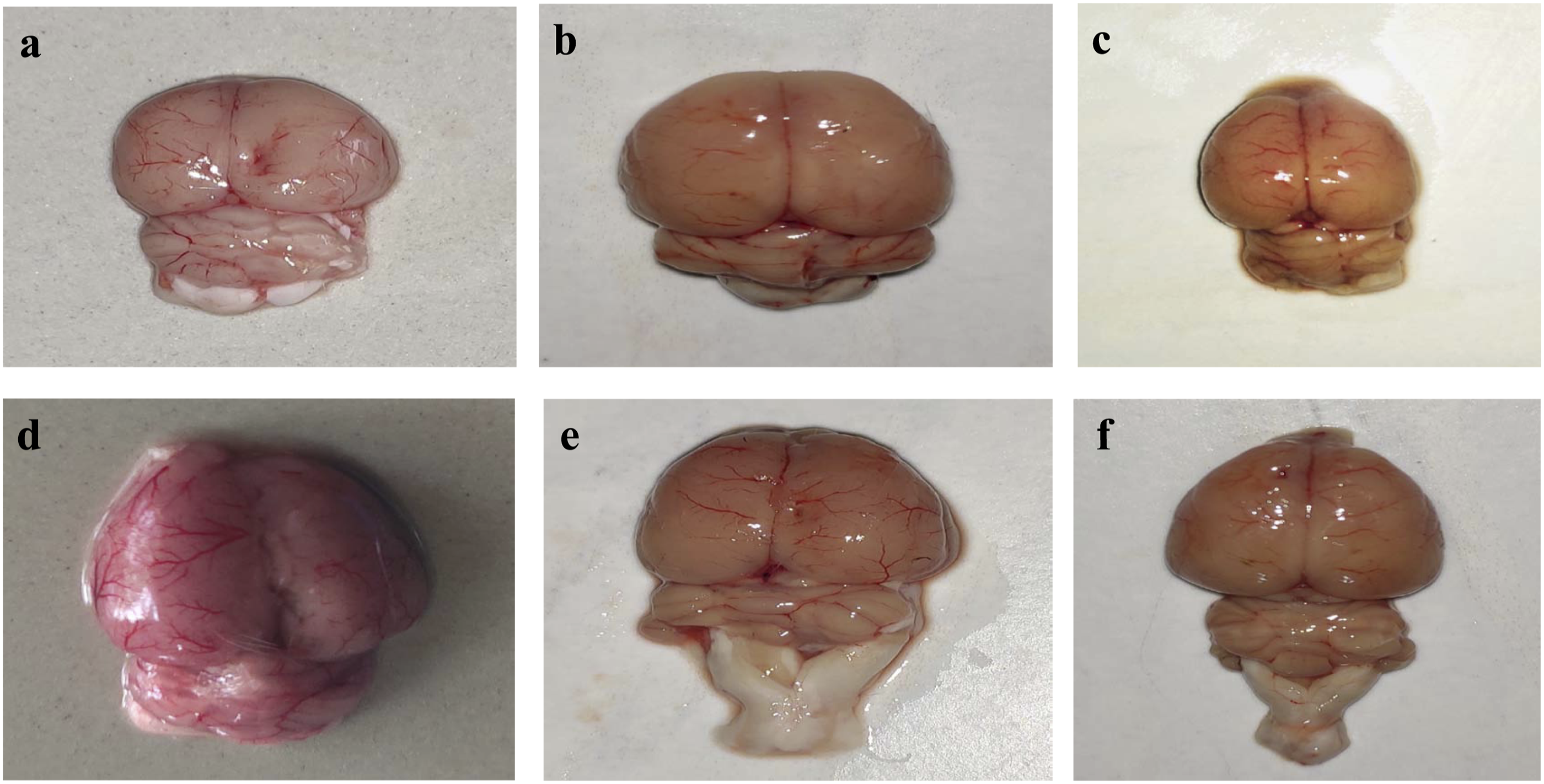
Improved pathological alteration in brain sections after prolonged treatment with apigenin
The normal control, vehicle control, and APG 80 perse-administered rat brain sections were seen to have a morphologically optimally shaped, clearly visible basal ganglia, cortex, and hippocampus tissue. MeHg-administered rat brain sections were affected by impaired, demyelinated, and aberrant tissues, resulting in a significant reduction in the hippocampus basal ganglia and cortex tissue compared to normal, vehicle, and APG-administered rats. A substantial improvement in demyelination and reduced pathological changes were observed in APG 40 mg/kg and APG 80 mg/kg treatment rats (Figure 10). Neuroprotective effect of apigenin in improvement of pathological alteration in brain sections in MeHg-induced neurotoxicity in adult rats: (a) (i) cerebral cortex, (ii) hippocampus, and (iii) basal ganglia; (b) vehicle control; (c) APG perse; (d) MeHg5; (e) MeHg + APG 40; and (f) MeHg + APG 80 (scale bar = 5 mm). Note: Yellow circles point to the site of the brain injury.
Reduced demyelination volume after prolonged treatment with apigenin
There was no substantial impact observed in the size of the demyelination area in comparison with normal, and vehicle control, and APG 80 perse groups. Prolong administration with MeHg considerably increases the demyelination area compared to the normal, vehicle, and APG 80 perse treatment groups. Moreover, prolonged treatment with APG 40 mg/kg and 80 mg/kg resulted in a remarkable reduction in the area of demyelination compared to groups that only received MeHg [one-way ANOVA: F (5,25) = 3.849, p < .01] (Figure 11). Neuroprotective effect of apigenin in reduction of demyelination volume in MeHg-induced neurotoxicity in adult rats. APG treatment decreased the demyelination volume and is expressed in mm3. Statistical analysis followed by one-way ANOVA (post hoc Tukey’s test). Values expressed as mean ± SEM(n = 6 rats per group). * p < .01 v/s normal control, vehicle control, and APG 80 perse; @ p < .01 v/s MeHg; @# p < .01 v/s MeHg + AP G40.
Discussion
This study investigated the neuroprotective efficacy of active phytoconstituent APG against MeHg-induced neurotoxicity in adult rats. Long-term treatment with APG 40 mg/kg and 80 mg/kg alone was shown to restore the multiple pathological neurocomplications associated with ALS. In addition, the model caused mental, cognitive, and behavioral deficits in some brain regions, including the cerebellum, cerebrum, hippocampus, and cortex, which were linked and matched to ALS-like neuropathological alterations.15,117,118
Oral administration of MeHg caused several behavioral and neuropathological changes. We tried to replicate this experimental model that mimics ALS-like neurobehavioral and biochemical defects. 15 The duration of our protocol was 42 days, with a low dose of MeHg. Impairment of motor function, loss of muscle control, typical memory, and cognitive impairment was reported with the administration of MeHg. Object-directed actions, coupled with increased neuroinflammation and oxidative stress markers, were also established during the research. 15 In the recent research, 21-day administration of APG significantly improved motor and behavioral dysfunction, along with a decrease in oxidative injury and neuroinflammation in MeHg-induced neurotoxicity in adult rats.
The body weight was considerably increased on days 35 and 42. APG 80 mg/kg enhanced locomotive performance significantly, indicating reduced glutamate levels during functional recovery and providing new insight into ALS treatment. APG 80 mg/kg administration significantly improved the escape latency in the Morris water maze, followed by the time-consuming target quadrant’s spatial navigation task. The forced swim test, open field test, locomotive behavior, and grip strength test also improved significantly from 28 to 42 days with persistent APG 80 mg/kg, showing its neuroprotective effect on brain functioning restoration.
We have also seen substantial increases in cellular and molecular markers (c-JNK and p38MAPK) and decreased demyelination marker (MBP) levels in the current research. The apoptotic marker level was also increased (caspase-3 and Bax) and decreased (Bcl-2) in MeHg-treated rats. However, when APG 40 mg/kg and 80 mg/kg were administered, the levels in the brain were significantly improved. Thus, we may assume that APG had a neuroprotective impact on reversing brain activity and a definite role for c-JNK and p38MAPK in MeHg-induced neurotoxicity in rats. In addition, the previous study also found that p38MAPK pharmacological inhibition decreased the level of inflammatory cytokines, 119 protected motor neurons 39 and neuritis degeneration, 43 and showed improvement within the physiological rate of axonal retrograde transport 40 in ALS. MeHg has a severe impact on various levels of brain neurotransmitters. In the diseased group, Ach, TBARS, and nitrite levels increased, while LDH, SOD, and GSH decreased significantly.120,121 These levels were further improved by long-term treatment of APG 40 mg/kg, and APG 80 mg/kg increased the functioning of the basal forebrain in the cholinergic system, resulting in cognitive improvements. Our research also confirmed that oral administration of MeHg increases the release of inflammatory mediators like TNF-α and IL-1β, which induce several neurobiological effects117,122,123 as well as improved significantly with the induction of APG 40 mg/kg and 80 mg/kg. Besides, APG also modulates glutamate level, severely increasing in the diseased group. The Ach, serotonin, and GABA levels decreased with the administration of MeHg but were found to increase with APG induction.
The above results demonstrated the protective effects of APG 40 mg/kg and 80 mg/kg on main pathological ALS pathways. Also, APG had a promising neuroprotective effect,62,124,125 anticancer activity,53,126 antiviral activity, 127 and antioxidant activity. 86 Long-term administration of APG 40 mg/kg and APG 80 mg/kg improved the neurological score for particular behavioral parameters. Also, it demonstrated a remarkable change in various neurochemical levels, showing its important neuroprotective function in reducing neuronal mortality and inflammation in patients.62,128 This study observed morphological and gross pathological alterations in rat brains following the generation of MeHg-induced neurotoxicity. Then, neurological functioning and the expression of several biomarkers were studied. The total area damaged by MeHg-associated demyelination was also assessed. The findings revealed no brain tissue injury in the normal, vehicle, or perse groups. On either side, the diseased group had significant demyelination in brain regions, which increased significantly with chronic APG treatment. The findings showed that APG has a neuroprotective effect, indicating that the animal model is well developed and manageable. Along with these, the volume of demyelination, basal ganglia, cortex, and hippocampus tissue grew significantly, indicating a positive response to our protocol drug.
However, the current findings are only associations in which the neuroprotective action of APG in MeHg-induced neurobehavioral and neurochemical alterations in ALS-treated rats was primarily investigated by altering c-JNK/p38MAPK-mediated signaling pathways and MBP restoration. Overexpression or deletion of the c-JNK and p38MAPK signaling pathways and additional molecular validation like immunohistopathology and immunoblotting are required to demonstrate the mechanism of action for future implications.
Conclusion
Based on our findings, an experimental model of MeHg-induced neurotoxicity in rats experienced neurochemical imbalance, demyelination, loss of muscular strength, motor weakness, memory, and cognitive failures, caused by cellular and molecular dysfunction. The c-JNK and p38MAPK pathways were involved. MeHg-treated rats demonstrated severe memory and cognitive loss and motor dysfunction. There was also upregulation of c-JNK and p38MAPK, demyelination, neuroinflammation, neuronal apoptosis, and oxidative stress. The long-term APG 40 mg/kg and APG 80 mg/kg treatment attenuates behavioral and neuropathological changes caused by MeHg administration.
Additionally, the level of the c-JNK and p38MAPK and MBP as well as apoptotic markers was observed to be significantly restored. Demyelination caused due to the chronic administration of MeHg was also marked to be improved in the rat brain tissue on the induction of the protocol drug. The levels of various neurotransmitters, neuroinflammatory markers, and oxidative stress markers were also improved in the study. The morphological and gross pathological changes in the entire brain and brain parts after the administration of MeHg showed a poor outcome, which was later improved when treated with the APG at both doses. Thus, APG may be a new therapeutic drug approach to ameliorate neuronal dysfunction, including changes in various neurochemicals. The study also confirms the involvement of the c-JNK and p38MAPK pathways in MeHg-induced neurotoxicity in rats. No such research has been performed until now regarding the neuroprotection effect of APG and its inhibition for c-JNK and p38MAPK in MeHg-treated rats. However, we have made no future investigations of Western blot immunohistopathology, which should be studied despite our research having just a few limitations.
Supplemental Material
sj-rar-1-het-10.1177_09603271221084276 – Supplemental Material for Protective effects of apigenin on methylmercury-induced behavioral/neurochemical abnormalities and neurotoxicity in rats
Supplemental Material, sj-rar-1-het-10.1177_09603271221084276 for Protective effects of apigenin on methylmercury-induced behavioral/neurochemical abnormalities and neurotoxicity in rats by Rajeshwar Kumar Yadav, Sidharth Mehan, Rakesh Sahu, Sumit Kumar, Andleeb Khan, Hafiz Antar Makeen and Mohammed Al Bratty in Human & Experimental Toxicology
Footnotes
Acknowledgments
The authors express their gratitude to Chairman, Mr. Parveen Garg, and Director, Dr. G. D. Gupta, ISF College of Pharmacy, Moga, Punjab, India, for their great vision and support.
Authors' contributions
All data were generated in-house, and no paper mill was used. All authors agree to be accountable for all aspects of work, ensuring integrity and accuracy.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
This work was supported by institutional grants from the Institutional Animal Ethics Committee (IAEC) with registration no. 816/PO/ReBiBt/S/04/CPCSEA as protocol no. ISFCP/IAEC/CPCSEA/Meeting No. 25/2019/Protocol No. 423 approved by RAB Committee, ISFCP, Moga, Punjab, India.
Ethical approval
All applicable institutional guidelines for the care and use of animals were followed.
Supplemental Material
Supplemental material for this article is available online.
Abbreviations
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.