
Abstract
Background
Circular RNA (circRNA) has been reported to regulate respiratory diseases. In the study, we aimed to elucidate the role of circ_0000157 in smoke-related chronic obstructive pulmonary disease (COPD) and the inner mechanism.
Methods
COPD-like cell injury was induced by treating human bronchial epithelioid cells (16HBE) with cigarette smoke extract (CSE). The expression of circ_0000157, miR-149-5p, bromodomain containing 4 (BRD4), BCL2-associated x protein (Bax) and B-cell lymphoma-2 (Bcl-2) was analyzed by quantitative real-time polymerase chain reaction (qRT-PCR) or Western blotting. Enzyme-linked immunosorbent assay was performed to detect interleukin-6 (IL-6) and tumor necrosis factor-α (TNF-α) levels. Malondialdehyde (MDA) production was detected by a lipid peroxidation MDA assay kit. Superoxide dismutase (SOD) activity was analyzed by a SOD activity assay kit.
Results
Circ_0000157 and BRD4 expression were upregulated, while miR-149-5p expression was downregulated in the blood of smokers with COPD and CSE-induced 16HBE cells compared with control groups. CSE treatment inhibited 16HBE cell proliferation and induced cell apoptosis, inflammation, and oxidative stress; however, these effects were remitted when circ_0000157 expression was decreased. In addition, circ_0000157 acted as a miR-149-5p sponge and regulated CSE-caused 16HBE cell damage by targeting miR-149-5p. The overexpression of BRD4, a target gene of miR-149-5p, attenuated the inhibitory effects of miR-149-5p introduction on CSE-induced cell damage. Further, circ_0000157 modulated BRD4 expression by associating with miR-149-5p in CSE-treated 16HBE cells.
Conclusion
Circ_0000157 knockdown ameliorated CSE-caused 16HBE cell damage by targeting the miR-149-5p/BRD4 pathway, providing a potential therapeutic strategy for clinic intervention in COPD.
Introduction
Chronic obstructive pulmonary disease (COPD) that accounts for about 2% of all inpatients in China is a progressive degenerative lung disease, accompanied by respiratory symptoms, such as shortness of breath and cough. 1 The risk factors of COPD include smoking, occupational dust and chemicals, genetics, intravenous drug use, connective tissue disorders and age. 2 Among these risk factors, smoking is considered the main one, and smoking-caused COPD cases represent over 80% of total COPD sufferers every year. 3 In terms of mechanism, cigarette smoking (CS) induces endothelial cell injury and later causes increased cell apoptosis and oxidative stress. 2 But the detailed mechanism has not been fully disclosed.
Circular RNA (circRNA) is a transcript possessing a covalently closed loop structure that is generated by back-splicing events and is widely expressed in eukaryotes. 4 As reported, circRNAs are dysregulated in many types of diseases, such as cardiovascular disease, 5 cancers 6 and respiratory diseases. 7 Several references described the roles of circRNAs in the pathogenesis of COPD. For example, circBbs9 aggravated particulate matter-caused lung inflammation by activating NLR family pyrin domain containing 3 (NLRP3) inflammasome in a COPD mouse model. 8 CircOSBPL2 depletion attenuated cigarette smoke extract (CSE)-caused human bronchial epithelial cell proliferation repression and apoptosis promotion. 9 Circ_0000157 is formed by head-to-tail splicing of the selectin L (SELL) and has been reported to be one of the 10 most upregulated circRNAs in COPD patients compared with control 10 ; however, the role of circ_0000157 in COPD remains unknown.
CircRNA can titrate microRNA (miRNA) to mediate gene expression, thus functioning in disease development. 11 MiRNA is a small noncoding RNA that plays a key part in the post-transcriptional modulation of mRNA. 12 In particular, miRNA enables mRNA to have vital regulatory roles in disease development. Previous data have indicated that smoking can affect miRNA expression, thus inducing COPD. 13 Considerable work demonstrated the pathophysiological roles of miRNA in COPD. For example, miR-195-5p repressed COPD development through interaction with sialic acid-binding immunoglobulin-like lectin1 (siglec1). 14 Ectopic expression of miR-145-5p reduced CSE-triggered apoptosis and inflammation through Kruppel-like 5 (KLF5). 15 Another miRNA, miR-149-5p, has been reported to participate in the progression of inflammation-related diseases, such as advanced atherosclerosis, 16 hepatic fibrosis 17 and osteoarthritis. 18 Nevertheless, its function in COPD has not been completely revealed.
Herein, we aimed to investigate the expression properties and regulatory role of circ_0000157 in smoke-related COPD using a CSE-induced human bronchial epithelioid cell (16HBE) model. In addition, we determined whether circ_0000157 bound to miR-149-5p and acted as a miR-149-5p sponge to increase the expression of miR-149-5p target gene, which resulted in CSE-induced 16HBE injury and thus contributed to COPD occurrence.
Materials and methods
Clinical samples
The whole blood samples were collected from 61 lung cancer sufferers diagnosed with solitary pulmonary tumors, including 19 non-smokers without COPD, 20 smokers without COPD and 22 smokers with COPD, at the Second Hospital of Hebei Medical University. Supernatant sera were harvested by centrifuging these samples at 1200 g for 12 min and saved at −80°C until RNA and protein isolation. All related patients signed the written informed consent for sample use. All participants had no other diseases. The work was performed under the permission of the Ethics Committee of the Second Hospital of Hebei Medical University (permission date: 20 October 2021).
CSE preparation
We prepared CSE according to the previous method. 9 In brief, reformative syringe-driven apparatus was used to obtain CSE from commercial cigarettes (Liqun Cigarette Factory, Hangzhou, China). The smoke was bubbled using 20 mL of Roswell Park Memorial Institute-1640 (RPMI-1640; Biosun, Shanghai, China). Large bacteria and particles were removed with 0.2 μm pore-size filters. The working concentrations of CSE were obtained by diluting the resulting suspension using phosphate-buffered saline (PBS; Millipore, Billerica, MA, USA).
Cell culture and CSE treatment
16HBE cells were provided by Otwo Biotech (Shenzhen, China) and cultured in RPMI-1640 (Biosun) added with 10% fetal bovine serum at 37°C in a biochemical incubator. For CSE-related assays, 16HBE cells were treated with working concentrations of CSE (0%, 1%, 2% and 3%) for 24 h, as instructed. 9
Cell transfection
Small interfering RNA of circ_0000157 (si-circ_0000157 5′-AGAAGTGGGGCAGGGAGAGTA-3′; binding to the back-splice junction sites), miR-149-5p mimics (5′-UCUGGCUCCGUGUCUUCACUCCC-3′), miR-149-5p inhibitors (anti-miR-149-5p 5′-GGGAGUGAAGACACGGAGCCAGA-3′) and the corresponding controls (si-NC, miR-NC and anti-miR-NC) were synthesized by Ribobio (Shanghai, China). Recombinant vector was generated with the use of coding sequences of bromodomain containing 4 (BRD4) and the empty vector (pcDNA 3.1(+); restriction enzyme cutting sites, BamH I and Xho I), and termed as BRD4. 16HBE cells were transfected with the above plasmids and oligonucleotides utilizing FuGENE6 (Roche, Basel, Switzerland).
Identification of groups
After 24 h of 2% of CSE treatment, 16HBE cells were transfected with si-circ_0000157, si-NC, anti-miR-149-5p, anti-miR-NC, miR-149-5p, miR-NC, BRD4 and pcDNA alone or jointly utilizing FuGENE6 (Roche), generating 2% CSE group, 2% CSE+si-NC group, 2% CSE+si-circ_0000157 group, 2% CSE+si-circ_0000157+anti-miR-NC group, 2% CSE+si-circ_0000157+anti-miR-149-5p group, 2% CSE+miR-NC group, 2% CSE+miR-149-5p group, 2% CSE+miR-149-5p+pcDNA group and 2% CSE+miR-149-5p+BRD4 group. 16HBE cells treated with 0% CSE were used as blank controls. Test groups and classification criteria are shown in Supplemental Table S1.
Cell counting kit-8 (CCK-8) assay
16HBE cells were seeded in 96-well plates and treated with various concentrations of CSE (0%, 1%, 2% and 3%) for 24 h. Then, si-circ_0000157, si-NC, anti-miR-149-5p, anti-miR-NC, miR-149-5p, miR-NC, BRD4 and pcDNA were transfected into the cells alone or jointly. After 48 h, the cells were exposed to 10 μL of CCK-8 reagent (Dojindo, Shanghai, China) for 4 h. At last, enzyme immunoassay analyzer (Bio-Rad, Hercules, CA, USA) was used to analyze the absorbance at 450 nm.
5-Ethynyl-2′-deoxyuridine (EdU) assay
DNA synthesis in 16HBE cells with various treatments was analyzed by EdU staining kit (Ribobio) as per the guidebook. In short, 16HBE cells were allowed to grow in 96-well plates and then incubated with EdU labeling media for 2 h in an incubator with 5% CO2, followed by treating with anti-EdU working solution. At last, EdU-positive cells from six fields of view were analyzed under confocal microscopy.
Flow cytometry analysis
16HBE cells with transfection and/or treatment were diluted in binding buffer (Solarbio, Beijing, China) with a concentration of 1 × 106 cells/mL. Then, the cells were incubated with Annexin V-FITC (Solarbio) and propidium iodide (PI; Solarbio) in the dark, lasting 10 min. At last, apoptotic cells were quantified with flow cytometry.
Western blotting assay
After RIPA lysis buffer treatment, the extracted proteins from cells and tissues were quantified using a spectrophotometer. Afterward, 20 μg of protein was loaded onto SurePAGE gels (Thermo Fisher, Waltham, MA, USA) with a SureLock Midi-Cell Electrophoresis System. The proteins were transferred onto nitrocellulose membranes and probed with the following primary antibodies, anti-BCL2-associated x protein (anti-Bax; Cat#PA5-11378; 1:2000; Thermo Fisher), anti-B-cell lymphoma-2 (anti-Bcl-2; Cat#PA5-27094; 1:2000; Thermo Fisher), anti-BRD4 (Cat#PA5-41550; 1:1000; Thermo Fisher) and anti-glyceraldehyde-3-phosphate dehydrogenase (anti-GAPDH; Cat#PA1-987; 1:3000; Thermo Fisher; serving as an internal reference). After washing using Tris-buffered saline, the membranes with incubated with secondary antibodies (Thermo Fisher). Blots were visualized using eyoECL Plus (Beyotime, Shanghai, China).
Enzyme-linked immunosorbent assay
Interleukin-6 (IL-6) and tumor necrosis factor-α (TNF-α) levels were analyzed using enzyme-linked immunosorbent assay (ELISA) kits (Beyotime) as per the guidebook. In brief, cell supernatant was harvested and added into each well of 96-well plates. After 2 h of incubation, the samples were incubated with biotinylated antibodies and Streptavidin. At last, enzyme immunoassay analyzer (Bio-Rad) was used to determine optical density at OD450.
Malondialdehyde (MDA) and Superoxide Dismutase (SOD)
For lipid peroxidation MDA assay, about 2 × 106 16HBE cells were harvested after treatment and/or cell transfection. The cells were washed using PBS and lysed using MDA lysis buffer (Abcam, Cambridge, MA, USA). Cell supernatant was collected by centrifugation and incubated with thiobarbituric acid (Abcam) at 95°C for 1 h, followed by analysis using a microplate reader (Thermo Fisher). For SOD activity assay, 16HBE cells were collected and lysed using lysis buffer (Abcam), followed by centrifugation to harvest cell supernatant. The following assay was performed inferring to the instruction of SOD activity assay kit (Abcam).
Quantitative real-time polymerase chain reaction (qRT-PCR)
Primer sequences used in qRT-PCR.

RNase R analysis
Total RNA (2 μg) was subjected to 20-min incubation in a humid incubator with or without RNase R (Geneseed, Guangzhou, China). Then, RNA was purified and circ_0000157 expression was analyzed by qRT-PCR. GAPDH served as a control gene.
Subcellular fractionation location assay
The separation of cytoplasmic and nuclear RNA in 16HBE cell samples was conducted as per the guidebook of PARIS Kit (Thermo Fisher). In short, the harvested cells were suspended in Cell Buffer, and then the cytoplasm of 16HBE cells was aspirated from the nuclear fractions. The cytoplasmic and nuclear RNA were analyzed by qRT-PCR.
Dual-luciferase reporter assay
Circular RNA interactome and starbase databases were performed to identify the complementary sites of circ_0000157 with miR-149-5p and BRD4. The wild-type (WT) sequences of circ_0000157 and BRD4 3′-untranslated region (3′UTR) containing the binding sites were obtained from TaKaRa Biotech, and later inserted into the pmirGLO Vector to generate WT-circ_0000157 and WT-BRD4 3′UTR. The complementary sites within circ_0000157 and BRD4 3′UTR were mutated by TaKaRa Biotech and mutant sequences were used to generate mutant plasmids, including MUT-circ_0000157 and MUT-BRD4 3′UTR. Subsequently, miR-149-5p mimics, miR-NC, and the above plasmids were co-transfected into 16HBE cells using FuGENE6 (Roche). Luciferase activity was assessed with Dual-Lucy Assay Kit (Solarbio) upon 48-h transfection.
RNA pull-down analysis
16HBE cells were transfected with biotin-labeled miR-149-5p (bio-miR-149-5p) and bio-miR-NC and then subjected to culture for 48 h. Then, the lysates, prepared using lysis buffer, were incubated with Streptavidin MagneSphere (Sigma, St. Louis, MO, USA) to capture the miR-149-5p-associated RNA complexes. At last, qRT-PCR analysis was conducted.
RNA immunoprecipitation (RIP) assay
As shown previously, 19 EZ-Magna RIP Kit (Millipore) was used to perform AGO2-RIP experiments in 16HBE cells with Ago2 and IgG antibodies (Abcam). In brief, cells were lysed by lysis buffer, and the lysates were then incubated with magnetic beads conjugated with the antibodies for 8 h. After being digested with proteinase K, the co-precipitated RNAs were analyzed by qRT-PCR.
Statistical analysis
Statistics analysis was carried out using GraphPad Prism software. Data were obtained from three independent duplicate tests and expressed as means ± standard deviations (SD). Distribution normality was evaluated using Shapiro-Wilk normality test. Significant differences between the two groups were determined using Student’s t-tests. Significant differences among three or more groups were performed using analysis of variance or Kruskal-Wallis test (used for nonparametric statistical analysis). p < 0.05 indicated statistical significance.
Results
CSE treatment induced 16HBE cell damage in a dose-dependent manner
The study first analyzed the effects of various doses of CSE (0%, 1%, 2% and 3%) on 16HBE cell processes. We found that CSE treatment dose-dependently inhibited 16HBE cell viability and proliferation (Figure 1(a) and (b)). In addition, CSE exposure induced 16HBE cell apoptosis in a dose-dependent manner, accompanied by an increase in Bax protein expression and a decrease in Bcl-2 expression (Figure 1(c)–(e)). As presented in Figure 1(f)–(h), CSE treatment dose-dependently increased IL-6, TNF-α and MDA levels and decreased SOD activity, suggesting the promoting effects of CSE on inflammation response and oxidative stress. Thus, these results suggested that CSE could induce COPD-like cell injuries. 2% of CSE was chosen for the following study as 50% cell viability under this concentration. The effects of various concentrations of CSE on 16HBE cell processes. (a)–(h) 16HBE cells were treated with 0%, 1%, 2% and 3% of CSE, and cell viability was analyzed by CCK-8 assay (a), cell proliferation by EdU assay (b), cell apoptosis by flow cytometry analysis (c), the protein expression of Bax and Bcl-2 by Western blotting analysis (d) and (e), the levels of IL-6 and TNF-α by ELISA (f), MDA production by lipid peroxidation MDA assay kit (g) and SOD activity by superoxide dismutase activity Assay kit (h). *p < 0.05, **p < 0.01, ***p < 0.001 and ****p < 0.0001.
Circ_0000157 expression was upregulated in the blood of COPD patients and CSE-induced 16HBE cells
Then, we analyzed circ_0000157 expression in the blood samples of COPD cases and CSE-treated 16HBE cells. As shown in Figure 2(a), circ_0000157 expression was higher in the blood of smokers and the highest in the blood of the smokers with COPD than in non-smokers. Moreover, the results showed that CSE treatment dose-dependently increased circ_0000157 expression (Figure 2(b)). Subsequently, the circular structure of circ_0000157 was confirmed using RNase R and Oligo(dT)18 primers. For instance, RNase R treatment significantly reduced GAPDH expression but not the expression of circ_0000157 (Figure 2(c)). Meanwhile, circ_0000157 amplified using random primers was more than that amplified using Oligo(dT)18 primers; however, there was no significant difference in GAPDH expression (Figure 2(d)). Further, the subcellular fractionation location assay showed that circ_0000157 was mainly located in the cytoplasm of 16HBE cells, as compared with the control groups (Figure 2(e)). These data demonstrated that COPD pathogenesis might involve circ_0000157. The expression of circ_0000157 in the blood of COPD patients and CSE-treated 16HBE cells. (a) Circ_0000157 expression was detected by qRT-PCR in the blood samples from non-smokers (N = 19), smokers (N = 20), and smokers with COPD (N = 22). (b) Circ_0000157 expression was examined by qRT-PCR in the 16HBE cells treated with 0%, 1%, 2%, and 3% CSE. (c) and (d) The circular structure of circ_0000157 was identified using RNase R, Oligo(dT)18 primers and random primers. (e) Subcellular fractionation location assay demonstrated that circ_0000157 was mainly located in the cytoplasm. **p < 0.01, ***p < 0.001 and ****p < 0.0001.
Circ_0000157 depletion protected 16HBE cells from CSE-induced damage
The study continued to analyze whether circ_0000157 participated in CSE-caused 16HBE cell injury. To determine this, we knocked down circ_0000157 expression in CSE-treated 16HBE cells. The data from Figure 3(a) first showed that CSE treatment increased circ_0000157 expression, whereas the effect was attenuated by decreasing circ_0000157 expression. Subsequently, CSE exposure induced inhibition of cell viability and cell proliferation, whereas these effects were remitted when circ_0000157 expression was reduced (Figure 3(b) and (c)). Meanwhile, circ_0000157 downregulation rescued the promoting effects of CSE on cell apoptosis and Bax expression and the inhibitory effect on Bcl-2 expression (Figure 3(d) and (e)). Comparatively, CSE treatment promoted IL-6, TNF-α and MDA production and inhibited SOD activity; however, these effects were relieved after circ_0000157 depletion (Figure 3(f)–(h)). To sum up, these findings manifested that circ_0000157 knockdown protected against CSE-caused 16HBE cell damage. Circ_0000157 knockdown protected against CSE-caused 16HBE cell damage. 16HBE cells were treated with 0% CSE, 2% CSE, 2% CSE+si-NC, or 2% CSE+si-circ_0000157, and circ_0000157 expression was analyzed by qRT-PCR (a), cell viability was analyzed by CCK-8 assay (b), cell proliferation by EdU assay (c), cell apoptosis by flow cytometry analysis (d), the protein expression of Bax and Bcl-2 by Western blotting analysis (e), the levels of IL-6 and TNF-α by ELISA (f), MDA production by lipid peroxidation MDA assay kit (g) and SOD activity by superoxide dismutase activity Assay kit (h). **p < 0.01, ***p < 0.001 and ****p < 0.0001.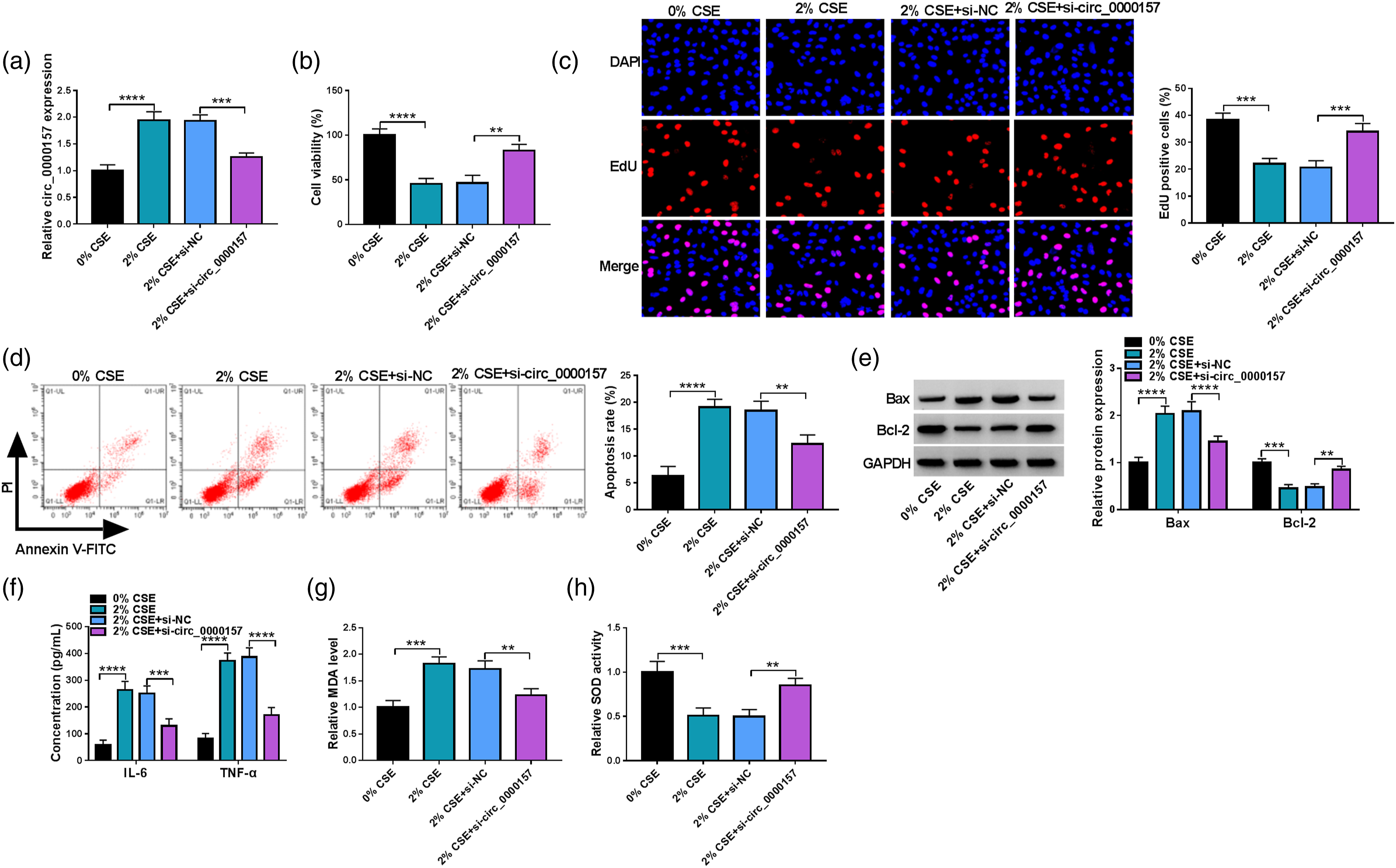
Circ_0000157 acted as a miR-149-5p sponge in 16HBE cells
Circular RNA interactome online database was used to identify the binding sites of circ_0000157 and miR-149-5p, and the results were shown in Figure 4(a). We then analyzed the efficiency of miR-149-5p mimics in increasing miR-149-5p expression using qRT-PCR. The results showed that miR-149-5p expression was significantly upregulated after transfection with miR-149-5p mimics compared with the control group (Figure 4(b)). Subsequently, dual-luciferase reporter assay showed that miR-149-5p overexpression reduced the luciferase activity of wild-type reporter plasmid of circ_0000157 but not the luciferase activity of mutant reporter plasmid (Figure 4(c)). In addition, biotinylated miR-149-5p could dramatically enrich circ_0000157 compared with biotinylated miR-NC (Figure 4(d)). RIP assay also showed that miR-149-5p and circ_0000157 expression were higher in the anti-Ago2 group than in the anti-IgG group (Figure 4(e)). Comparatively, miR-149-5p expression was lower in the blood of smokers without COPD and was the lowest in the blood of smokers with COPD than in the blood of non-smokers (Figure 4(f)). As revealed by Spearman correlation analysis, circ_0000157 expression was negatively correlated with miR-149-5p in the blood of smokers with COPD (Figure 4(g)). Further, CSE treatment dose-dependently reduced miR-149-5p expression in 16HBE cells (Figure 4(h)). In a word, the above data demonstrated that circ_0000157 bound to miR-149-5p in 16HBE cells. Circ_0000157 interacted with miR-149-5p in 16HBE cells. (a) The schematic illustration showed the complementary sites of circ_0000157 with miR-149-5p. (b) The efficiency of miR-149-5p overexpression was determined by qRT-PCR in 16HBE cells. (c)–(e) Dual-luciferase reporter assay, RNA pull-down assay, and RIP assay were performed to analyze the regulatory relationship of circ_0000157 and miR-149-5p in 16HBE cells. (f) MiR-149-5p expression was detected by qRT-PCR in the blood samples from non-smokers (N = 19), smokers (N = 20), and smokers with COPD (N = 22). (g) Spearman correlation analysis was performed to analyze the correlation of miR-149-5p and circ_0000157 expression in the blood samples of smokers with COPD. (h) MiR-149-5p expression was detected by qRT-PCR in the 16HBE cells treated with 0%, 1%, 2% or 3% CSE. **p < 0.01, ***p < 0.001 and ****p < 0.0001.
Circ_0000157 modulated CSE-induced 16HBE cell injury by interacting with miR-149-5p
Given the interaction of circ_0000157 and miR-149-5p, we further investigated whether miR-149-5p was involved in the regulation of circ_0000157 on CSE-induced 16HBE cell dysfunction. The results showed that CSE treatment-induced decrease in miR-149-5p expression was remitted after circ_0000157 depletion, whereas the effect was relieved by reducing miR-149-5p expression (Figure 5(a)). Subsequently, CSE treatment-induced repression on cell viability and cell proliferation was restored by decreasing circ_0000157 expression, but the effect was rescued when miR-149-5p expression was downregulated (Figure 5(b) and (c)). Consistently, miR-149-5p depletion remitted circ_0000157 knockdown-induced inhibition on cell apoptosis and the dysregulation of Bax and Bcl-2 expression in CSE-treated 16HBE cells (Figure 5(d)–(f)). Further, circ_0000157 absence attenuated the promoting effects of CSE treatment on IL-6, TNF-α and MDA production and the inhibitory effect on SOD activity; however, these effects were relieved by decreasing miR-149-5p expression (Figure 5(g)–(i)). Thus, the above evidence demonstrated that the circ_0000157/miR-149-5p pathway regulated CSE-induced 16HBE cell injury. Circ_0000157 modulated CSE-induced 16HBE cell injury by interacting with miR-149-5p. 16HBE cells were divided into six groups, including 0% CSE group, 2% CSE group, 2% CSE+si-NC group, 2% CSE+si-circ_0000157 group, 2% CSE+si-circ_0000157+anti-miR-NC group and 2% CSE+si-circ_0000157+anti-miR-149-5p group. (a) MiR-149-5p expression was analyzed by qRT-PCR. (b) Cell viability was detected by CCK-8 assay. (c) Cell proliferation was investigated by EdU assay. (d) Cell apoptosis was assessed by flow cytometry analysis. (e) and (f) Bax and Bcl-2 protein expression were checked by Western blotting analysis. (g) ELISA assay was used to detect IL-6 and TNF-α levels. (h) MDA production was determined by lipid peroxidation MDA assay kit. (i) SOD activity was analyzed with superoxide dismutase activity Assay kit. *p < 0.05, **p < 0.01, ***p < 0.001 and ****p < 0.0001.
BRD4 was identified as a target gene of miR-149-5p in 16HBE cells
The study continued to search for the target mRNA of miR-149-5p through the starbase online database. As shown in Figure 6(a), BRD4 possessed the binding sites of miR-149-5p. Also, the luciferase activity of wild-type reporter plasmid of BRD4 3′UTR was significantly inhibited by increasing miR-149-5p expression, while that of mutant reporter plasmid had no response to miR-149-5p introduction (Figure 6(b)). In addition, we observed that BRD4 expression was higher in the bio-miR-149-5p group than in the bio-miR-NC group (Figure 6(c)). RIP assay showed that both miR-149-5p and BRD4 were significantly enriched by anti-Ago2 compared with anti-IgG (Figure 6(d)). Based on the above results, BRD4 was employed as a follow-up study subject. Comparatively, BRD4 expression was higher in the blood samples of smokers and the highest in the blood samples of smokers with COPD (Figure 6(e)). BRD4 expression was negatively correlated with miR-149-5p in the blood of smokers with COPD, as presented in Figure 6(f). Consistently, CSE treatment led to upregulation of BRD4 expression in a concentration-dependent manner (Figure 6(g)). Further, circ_0000157 depletion reduced BRD4 production in CSE-induced 16HBE cells, whereas the effect was remitted by decreasing miR-149-5p expression (Figure 6(h)). MiR-149-5p targeted BRD4 in 16HBE cells. (a) The schematic illustration showed the complementary sites of miR-149-5p with BRD4. (b)–(d) The association of miR-149-5p and BRD4 was identified by dual-luciferase reporter assay, RNA pull-down assay, and RIP assay. (e) qRT-PCR was performed to detect BRD4 expression in the blood of non-smokers (N = 19), smokers (N = 20), and smokers with COPD (N = 22). (f) Spearman correlation analysis was carried out to assess the correlation of miR-149-5p and BRD4 expression in the blood samples of smokers with COPD. (g) BRD4 protein expression was detected by Western blotting in the 16HBE cells treated with 0%, 1%, 2% or 3% CSE. (h) The effects of circ_0000157 depletion and miR-149-5p absence on BRD4 expression were analyzed by Western blotting in CSE-treated 16HBE cells. **p < 0.01, ***p < 0.001 and ****p < 0.0001.
BRD4 overexpression attenuated the effects of miR-149-5p on CSE-induced 16HBE cell damage
The study further analyzed whether the regulation of miR-149-5p on CSE-caused 16HBE cell injury involved BRD4. To validate this, we overexpressed miR-149-5p and BRD4 in CSE-induced 16HBE cells. The results showed that miR-149-5p mimics downregulated BRD4 expression in CSE-induced cells, whereas the effect was reversed after BRD4 overexpression (Figure 7(a)). Subsequently, miR-149-5p introduction promoted cell viability and cell proliferation but repressed cell apoptosis under CSE treatment; however, these effects were remitted by increasing BRD4 expression (Figure 7(b)–(d)). In addition, the decreased Bax expression and increased Bcl-2 expression by miR-149-5p overexpression were attenuated when BRD4 expression was upregulated in CSE-induced 16HBE cells (Figure 7(e) and (f)). Further, we observed that miR-149-5p mimics inhibited IL-6, TNF-α, and MDA production and improved SOD activity in the cells, while the ectopic BRD4 expression relieved these effects (Figure 7(g)–(i)). Thus, these results demonstrated that the miR-149-5p/BRD4 pathway modulated CSE-induced 16HBE cell damage. MiR-149-5p regulated CSE-induced 16HBE cell injury by interacting with BRD4. (a)–(i) 16HBE cells were assigned to six groups, including 0% CSE group, 2% CSE group, 2% CSE+miR-NC group, 2% CSE+miR-149-5p group, 2% CSE+miR-149-5p+pcDNA group and 2% CSE+miR-149-5p+BRD4 group, BRD4 protein expression was analyzed by Western blotting analysis (a), cell viability by CCK-8 assay (b), cell proliferation by EdU assay (c), cell apoptosis by flow cytometry analysis (d), the protein expression of Bax and Bcl-2 by Western blotting analysis (e) and (f), the levels of IL-6 and TNF-α by ELISA (g), MDA production by lipid peroxidation MDA assay kit (h) and SOD activity by superoxide dismutase activity Assay kit (i). *p < 0.05, **p < 0.01, ***p < 0.001 and ****p < 0.0001.
Discussion
In recent years, more and more studies are performed to investigate the pathophysiological functions of circRNA in inflammation-related diseases, such as renal ischemia-reperfusion injury 20 and lung inflammation. 8 The present work was conducted to analyze circ_0000157 role in the pathogenesis of COPD and the underneath mechanism. Our data showed that circ_0000157 depletion reduced BRD4 expression to protect against CSE-induced 16HBE cell damage through miR-149-5p. Our evidence points to the intrinsic functions of circ_0000157 in COPD.
Multiple studies indicated the involvement of circRNAs in COPD development. 10 However, the data were absent, regarding circ_0000157 function in COPD, especially in smoke-related COPD. In this study, we found that circ_0000157 was upregulated in the blood of smokers with COPD compared with smokers without COPD and non-smokers without COPD. Moreover, qRT-PCR analysis revealed circ_0000157 expression tended to be increased in CSE-exposed 16HBE cells in a concentration-dependent manner. Given that circRNA was more resistant to RNA exonuclease degradation, 21 we then analyzed circRNA stability using RNase R. Our results revealed that circ_0000157 was able to resist exonuclease digestion. Oligo (dT)18 primers could only amplify RNA containing poly (A) tails, while random primers could amplify all RNA. 22 Our data showed that circ_0000157 amplified using random primers was more than that amplified using Oligo(dT)18 primers, which indicated that circ_0000157 contained poly (A) tails. The above results demonstrated that circ_0000157 was a circular RNA and COPD occurrence might involve circ_0000157 upregulation.
Rescue experiments demonstrated that circ_0000157 absence remitted the inhibitory effects of CSE on 16HBE cell viability and cell proliferation and the promoting effects on IL-6 and TNF-α production. The Bcl-2 protein family regulates the mitochondrial pathway and is divided into pro-survival (such as Bcl-2) and pro-apoptotic proteins (such as Bax).23,24 In this work, Bcl-2 and Bax were used to evaluate cell apoptosis. We found that CSE treatment increased cell apoptotic rate and Bax protein expression and decreased Bcl-2 protein expression, whereas these effects were attenuated after circ_0000157 silencing. Oxidative stress can increase bronchial hyperresponsiveness, destruct airway epithelial cells, and impair antiprotease function and surfactant, thus considered as a major factor driving COPD. 25 Oxidative damage marker MDA and antioxidant defense marker SOD were employed in this study. Herein, we found that CSE treatment increased MDA production and decreased SOD activity; however, these effects were rescued when circ_0000157 expression was downregulated. Thus, our data demonstrated that circ_0000157 repression could prevent CSE-induced lung cell injury.
In addition, our data showed that circ_0000157 was located in the cytoplasm of 16HBE cells. Cytoplastic circRNA can regulate disease progression by combining with miRNA.26,27 To determine the detailed mechanism of circ_0000157 actions in CSE-induced cell damage, we searched for circ_0000157-associated miRNAs and identified miR-149-5p as its target miRNA. Early work has demonstrated that miR-149-5p combines with Notch2 to inhibit oxygen and glucose deprivation-induced IL-6 and TNF-α production and cell apoptosis. 28 Fu et al. 29 explained that miR-149-5p inhibitors attenuated circ_0008956 depletion-induced protective effects on IL-1β-exposed chondrocytes. Additionally, long noncoding RNA (lncRNA) IL6-AS1 interacted with miR-149-5p to promote IL-6 transcription, further increasing H3K4 histone methylation and H3K27 histone acetylation in COPD. 30 Herein, we confirmed the low expression of miR-149-5p in smokers with COPD and CSE-exposed 16HBE cells for the first time. Moreover, miR-149-5p could relieve CSE-caused inhibitory effect on cell proliferation and promoting effects on cell apoptosis, cell inflammation and oxidative stress. Functional analysis assays further manifested that circ_0000157 mediated CSE-induced lung cell damage through miR-149-5p.
CircRNA can function as a miRNA sponge to counteract miRNA-mediated mRNA expression inhibition. 31 Next, we searched for miR-149-5p-binding mRNA and identified BRD4 as its target gene. BRD4 is a member of the Bromodomains and Extra-terminal family and is an epigenetic regulator through interaction with acetylated histones. 32 In addition, the protein mediated inflammatory gene NF-κB expression by combining with acetylated RELA. 33 Previous references have revealed that BRD4 interacts with circANKRD11/miR-145-5p axis 34 and the lncRNA MIR155HG/miR-128-5p pathway 35 to increase CSE-caused HPMEC damage in COPD. Meanwhile, Zheng et al. 9 indicated that BRD4 could improve CSE-evoked human bronchial epithelial cell inflammation and oxidative stress promotion. Consistently, the present work revealed the high expression of BRD4 in the blood of smokers with COPD and CSE-exposed 16HBE cells. BRD4 re-expression decreased CSE-treated 16HBE cell proliferation and increased cell apoptosis, inflammation and oxidative stress. Further, circ_0000157 depletion repressed BRD4 production due to miR-149-5p downregulation in CSE-induced 16HBE cells. Thus, circ_0000157 regulated CSE-induced 16HBE cell injury by the miR-149-5p/BRD4 pathway. As revealed by a mouse model assay, BRD4 inhibitors protected against COPD by inhibiting the activation of the nuclear factor kappa B (NF-κB) pathway. 36 We proposed that circ_0000157 activated the miR-149-5p/BRD4 axis to mediate CSE-induced 16HBE cell damage through the NF-κB pathway.
Our study demonstrated the regulatory function of the circ_0000157/miR-149-5p/BRD4 pathway in COPD using a CSE-induced COPD cell model. However, two limitations should be considered in the present investigation. Firstly, all experiments were conducted on clinical COPD samples or CSE-induced cell model, and in vivo assay was absent. Additionally, the miR-149-5p/BRD4 pathway might be just one of the regulatory mechanisms related to circ_0000157-medicated COPD development, and more mechanisms should be investigated in the future.
Taken together, the present study confirmed that circ_0000157 depletion had a protective action against CSE-caused lung cell damage, and the detailed mechanism of the action was mediated by the circ_0000157/miR-149-5p/BRD4 axis. Thus, inhibiting circ_0000157 expression may be a reliable therapeutic approach to COPD.
Supplemental Material
Supplemental Material - Circular RNA 0000157 depletion protects human bronchial epithelioid cells from cigarette smoke extract-induced human bronchial epithelioid cell injury through the microRNA-149-5p/bromodomain containing 4 pathway
Supplemental Material for Circular RNA 0000157 depletion protects human bronchial epithelioid cells from cigarette smoke extract-induced human bronchial epithelioid cell injury through the microRNA-149-5p/bromodomain containing 4 pathway by B Song, S Wu, L Ye, Z Jing, and J Cao in Human & Experimental Toxicology
Footnotes
Author contributions
Beibei Song designed and performed the research; Siyu Wu, Liyun Ye, Zeng Jing and Jing Cao analyzed the data; Beibei Song wrote the manuscript. All authors read and approved the final manuscript.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Ethical approval
This study was permitted by the Ethics Committee of the Second Hospital of Hebei Medical University.
Informed consent
Written informed consents were obtained from all participants.
Supplemental Material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.