
Abstract
Background
MicroRNA-3612 (miR-3612) is considered a tumor suppressor in different cancers. Nonetheless, its function in nasopharyngeal carcinoma (NPC) has yet to be uncovered.
Methods
NPC cells and tissues were tested by means of reverse transcription quantitative polymerase chain reaction (RT-qPCR) analysis and western blotting to quantify the expressions of miR-3612 and Thrombospondin 1 (THBS1). Cell Counting Kit-8 (CCK-8) and scratch experiments were carried out to evaluate the migration and proliferation of NPC cells. NPC cell adhesion was also assessed. The predicted interaction of miR-3612 with THBS1 was verified by means of a luciferase reporter assay. In vivo experiments were also conducted to examine how miR-3612 overexpression affects in vivo tumorigenicity. Lastly, phosphatidylinositol 3-kinase (PI3K)/AKT signaling pathway status was assessed via western blotting.
Results
MiR-3612 was downregulated in NPC cells and tissues, whereas THBS1 expression showed an opposite trend. The MiR-3612 mimic inhibited the NPC cell proliferation, adhesion, and migration and also inactivated the PI3K/AKT signaling pathway. Furthermore, miR-3612 mimic also hampered NPC tumorigenesis in vivo. MiR-3612 targeted THBS1 and downregulated THBS1 expression. THBS1 offset the miR-3612-overexpression-induced repression of the migration, adhesion, and proliferation of NPC cells via the activation of the PI3K/AKT pathway.
Conclusion
MiR-3612 retarded NPC cell migration, adhesion, and proliferation by targeting THBS1 and inactivating the PI3K/AKT signaling pathway. This provides a novel therapeutic approach for NPC intervention.
Introduction
Nasopharyngeal Carcinoma (NPC), mainly present in the epithelium of the nasopharynx, was characterized by significant geographical variation in terms of incidence. Some existing epidemiological studies about NPC in China show an incidence from 15 to 50 per 100,000. 1 The burden of the disorder rises with genetic predisposition and Epstein Barr virus (EBV) infection.2,3 Due to its imperceptible symptoms, over 75% of NPC patients were first presented at an advanced stage. 3 Despite the great advancements in multimodality treatments for NPC, they are not beneficial for patients at the advanced stages of NPC. 4 Hence, investigating the mechanism that drives NPC progression became the major focus of this study.
MicroRNAs (miRNAs) are ∼22-nt, single stranded, non-coding RNAs. The functional importance of miRNA is evident in the normal functionalities of eukaryotic cells. MiRNA dysregulation plays a significant role in various disorders, including tumorigenesis. In NPC, miR-25 activates the Wnt/β-catenin signaling pathway and favors stepwise the malignant phenotypes of NPC cells. 5 MiR106a-5p manifests a tight association with the clinicopathologic parameters of NPC patients. 6 MiR-182-5p has been reported to promote radioresistance in NPC in vitro. 7 As for miR-3612, it has an anti-tumor role which was reported in various cancers, including melanoma, 8 esophageal squamous cell carcinoma, 9 and bladder cancer. 10 Nonetheless, the miR-3612s role in NPC requires elucidation.
Thrombospondin 1 (THBS1), also named as THBS, THBS-1, TSP, TSP-1, and TSP1, is a disulfide-linked homotrimeric protein subunit. It acts as a matricellular protein involved in cell–cell and cell–matrix interactions. Accumulating investigations attest to the critical biofunction of THBS1 in angiogenesis and tumorigenesis. 11 A high THBS1 expression is critical for the oncogenic activation of the hippo signaling pathway in breast cancer. 12 The pro-tumorigenic role of THBS1 was partially achieved in cutaneous T-cell lymphoma, in vivo and in vitro, through the regulation of AKT phosphorylation. 13 The hyperactivation of the PI3K/Akt/mTOR cascade in gastric cancer occurs as a response to THBS1 overexpression. 14 In lung cancer, however, THBS1 is under expressed in lung cancer and executes a tumor-suppressive role. 15 How THBS1 influences NPC progression is currently obscure.
Generally, miRNAs may silence mRNA expression by recognizing their target genes. 16 Through bioinformatics analyses, we found that miR-3612 targeted THBS1. In view of that, we planned to dissect the mechanism of the miR-3612/THBS1 axis that drives NPC progression. Our findings may put forward the therapeutic manipulation of the miR-3612/THBS1 axis as a candidate approach for NPC intervention.
Methods
Human tissue specimens
Thirty-four (34) sets of NPC and their adjacent uncancerous tissue specimens were gathered from NPC patients that never received any intervention for NPC. The ethical committee of our hospital authorized our research. Each patient provided an informed consent. All clinical specimens were maintained at −80°C prior the reverse transcription quantitative polymerase chain reaction (RT-qPCR) analysis.
Cell culture
Immortalized nasopharynx cells (NP69) and human NPC cells (NPC/HK1 and C666-1) were bought from the American Type Culture Collection. All the cell lines were authenticated using STR profiles. An RPMI-1640 medium (ThermoFisher, USA) with added 10% FBS (Thermo Fisher, USA) was utilized in cultivating the cells, and the cultures were maintained in an incubator with 5% CO2 at 37°C.
Cell transfection
MiR-3612 mimic (mimic), mimic-NC (NC), miR-3612 inhibitor (inhibitor), pCDNA-THBS1 vector for THBS1 overexpression (THBS1-OE), and its empty vector were all purchased from Gema (Shanghai, China). As described in the protocol, a Lipofectamine 3000 (Invitrogen, USA) was used in introducing the vectors into 80% confluent C666-1 and NPC/HK1 cells. After 48 h passed, the effectiveness of the transfection was verified by RT-qPCR.
RT-qPCR
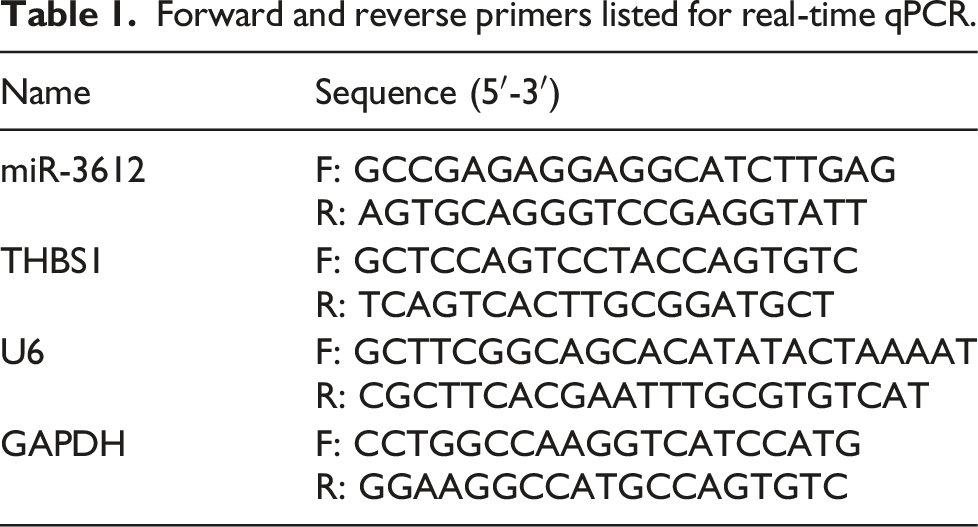
Forward and reverse primers listed for real-time qPCR.
Western blot
ProteoPre Total Extraction Kit (Merck, USA) was employed in the extraction of protein from NPC cells. After being assessed for concentration with a bicinchoninic acid kit (Merck, USA), the prepared protein samples (20 μg) were loaded on a 10% sulfate-polyacrylamidegel electrophoresis (SDS-PAGE) gel and then resolved at constant 60 V. The proteins were subsequently wet-transferred onto nitrocellulose. After their 1 h exposure to a blocking buffer at room temperature, the membranes were further incubated at 4°C with antibodies. On the following day, horseradish peroxidase (HRP)-conjugated goat anti-rabbit secondary antibody (ab202272, ab94820, 1:5,000, abcam) was utilized to blot the signals on membranes for 1 h at room temperature. The ECL Western Blotting Detection System (Fisher Scientific, USA) was then applied for the visualization of the blots. The primary antibodies used were the following: PI3K (ab191606, 1:1000, Abcam), p-PI3K (ab32089, 1:1000, Abcam), AKT (b283852, 1:1000, Abcam), p-AKT (ab204872, 1:1000, Abcam), THBS1 (sc-59,887, 1:1000, Santa Cruz), and GAPDH (ab8245, 1:1000, abcam).
Cell Counting Kit-8 (CCK-8) assays
For cell proliferation assessment, 2 × 103 NPC/HK1 and C666-1 cells were cultivated in each well of a 96-well culture plate. Both cells were treated with 10 μL CCK-8 reagent (Beyotime, China) at the indicated periods (0 h, 24 h, 48 h, and 72 h). Forty-eight hours after, the OD450 reading in each hole was taken with a microplate reader (Tecan, USA).
Scratch wound assays
After monolayers of NPC/HK1 and C666-1 cells formed on the 6-well plates, a 200 μL pipette tip was utilized in creating a vertical wound gap on the monolayer. Afterward, the detached cells were removed. At 0 and 48 h, an inverted microscope was utilized in capturing images of the wound closure. The image processing program ImageJ 1.43 (NIH, USA) was then utilized to evaluate the scratch gaps. Relative scratch = [(scratch width at 0 h – scratch width at 48 h) ÷ scratch width at 0 h × 100 %].
Cell adhesion assay
A total of 3 × 105 NPC/HK1 and C666-1 cells were grown in 96-well plates percolated with extracellular matrix (ECM). After incubation for 90 min, the cells were treated by 10 μl 3‐(4, 5‐dimethylthiazol‐2‐yl)‐2, 5‐diphenyltetrazolium bromide (MTT) (5 mg/mL) solution (ThermoFisher, USA). 4 h later, 100 μL DMSO (Promega, USA) were added for additional 10 min cultivation. Finally, the OD values (570 nm) of the wells were detected by a plate reader (ThermoFisher, USA).
Bioinformatic analysis
GSE12452 from GEO DataSets was used to screen the upregulated genes in NPC samples with the criteria of adj. p < 0.05 and logFC ≥1. TargetScan (https://www.targetscan.org/vert_71/) was applied to screen the target genes of miR-3612. The common genes from TargetScan and GSE12452 were identify by Venny 2.1.0 (https://bioinfogp.cnb.csic.es/tools/venny/). KEGG enrichment was analyzed by STRING (https://cn.string-db.org/).
Luciferase reporter assays
The wild-type (WT) miR-3612 binding sequence of THBS1 3′UTR and its respective mutant (MUT) sequences were individually inserted into pEZX-MT01 vectors with fLuc (GeneCopoeia, China) to produce the following recombinant vectors: THBS1 3′UTR-WT, THBS1 3′UTR-MUT1, THBS1 3′UTR-MUT2, THBS1 3′UTR-MUT3, THBS1 3′UTR-Co-MUT. These generated vectors, coupled with a miR-3612 mimic or mimic NC, were simultaneously introduced into the NPC/HK1 and C666-1 cells. Forty-eight hours later, a Luc-Pair Duo-Luciferase Assay Kit 2.0 (GeneCopoeia Inc. China) was utilized, as instructed, in the detection of the luciferase activity.
In vivo assays
Nude mice aged 5-6 weeks old were bought from Wuhan University Center for Animal Experiment/Animal Biosafety Level III laboratory (ABSL-III lab) (Wuhan, Hubei, China). All mice were bred under sterile specific-pathogen-free (SPF) environment and subjected to a light/dark cycle (12/12 h). Each mouse was subcutaneously injected with 1 × 103 C666-1 cells stably infected with agomiR-3612 or agomiR-NC. The volumes of the tumors were recorded every four days after modeling. Seven weeks later, the mice were euthanized after the administration of CO2, and the resected tumors were weighed.
Statistical analysis
The data were analyzed in GraphPad Prism 9 (Prism, USA) and were presented as the mean ± standard deviation (SD). Outcomes from two groups were compared by applying a paired t-test. Meanwhile, in comparing outcomes from more than two groups, One-way analysis of variance (ANOVA) accompanied with Bonferroni test was applied. Pearson correlation coefficient was adopted to test if the expressions of miR-3612 and THBS1 were correlated with each other in NPC tissues. p-values <0.05 were deemed significant.
Results
Overexpressing miR-3612 impedes NPC tumorigenesis in vitro by blunting the PI3K/AKT signaling pathway
Since miR-3612 reportedly has a role in various cancers,8,18 we carried out RT-qPCR analysis to ascertain its expression status in NPC cells and tissues. The NPC tissues (Figure 1(a)) and cells (Figure 1(b)) clearly exhibited a downregulation of miR-3612. The NPC/HK1 and C666-1 cells especially manifested approximately a 2-fold reduction in miR-3612 levels when compared to that in the NP69 cells. In view of this, we proceeded to explore the impact of miR-3612 dysregulation on NPC cell behaviors by introducing the miR-3612 mimic into the NPC/HK1 and C666-1 cells. As shown in Figure 1(c), the exogenous expression of miR-3612 effected by the miR-3612 mimic transfection led to about a 5-fold rise in miR-3612 expression among the NPC/HK1 and C666-1 cells. This is indicative of the successful transfection. The following proliferation analysis showed that miR-3612 overexpression impaired NPC cell proliferation (Figure 1(d)). Moreover, a declined adhesion was observed in NPC/HK1 and C666-1 cells overexpressing miR-3612 (Figure 1(e)). A similar trend was observed when a lower migration rate was detected among miR-3612-overexpressing NPC cells than in the control cells (Figure 1(f)). The PI3K/AKT signaling pathway is characterized as a pathway critical to the regulation of tumor cell oncogenicity.19,20 Hence, we also determined the influence of miR-3612 on this oncogenic pathway. Unsurprisingly, miR-3612 amplification robustly reduced the expressions of p-PI3K and p-AKT (Figure 1(g)). Therefore, these suggest that miR-3612 overexpression repressed the migration, adhesion, and proliferation of NPC cells throughs the PI3K/AKT signaling pathway. MiR-3612 overexpression impedes NPC cell migration, adhesion, and proliferation by inhibiting the PI3K/AKT signaling pathway. (a) RT-qPCR analysis of miR-3612 expression in NPC tumors and matching uncancerous tissues. (b) RT-qPCR analysis of miR-3612 expression in NP69 cells and NPC cells (NPC/HK1 and C666-1). **p < 0.001 vs. NP69. (c) MiR-3612 expression in NPC/HK1 and C666-1 cells at 48 h post-transfection of miR-3612 mimic or mimic NC was analyzed via RT-qPCR. **p < 0.001 vs. NC. (d) CCK-8 analysis of transfected NPC/HK1 and C666-1 cells carrying mimic NC or miR-3612 mimic. **p < 0.001 vs. NC. (e) Tumor cell adhesion was assessed in transfected NPC/HK1 and C666-1 cells. **p < 0.001 vs. NC. (f) Scratch wound assays was carried out to assess the migration of NPC cells with a miR-3612 mimic or mimic NC. **p < 0.001 vs. NC. (g) Western blotting analysis of the critical effectors in PI3K/AKT signaling pathway in NPC/HK1 and C666-1 cells transfected with miR-3612 mimic or mimic NC. **p < 0.001 vs. NC.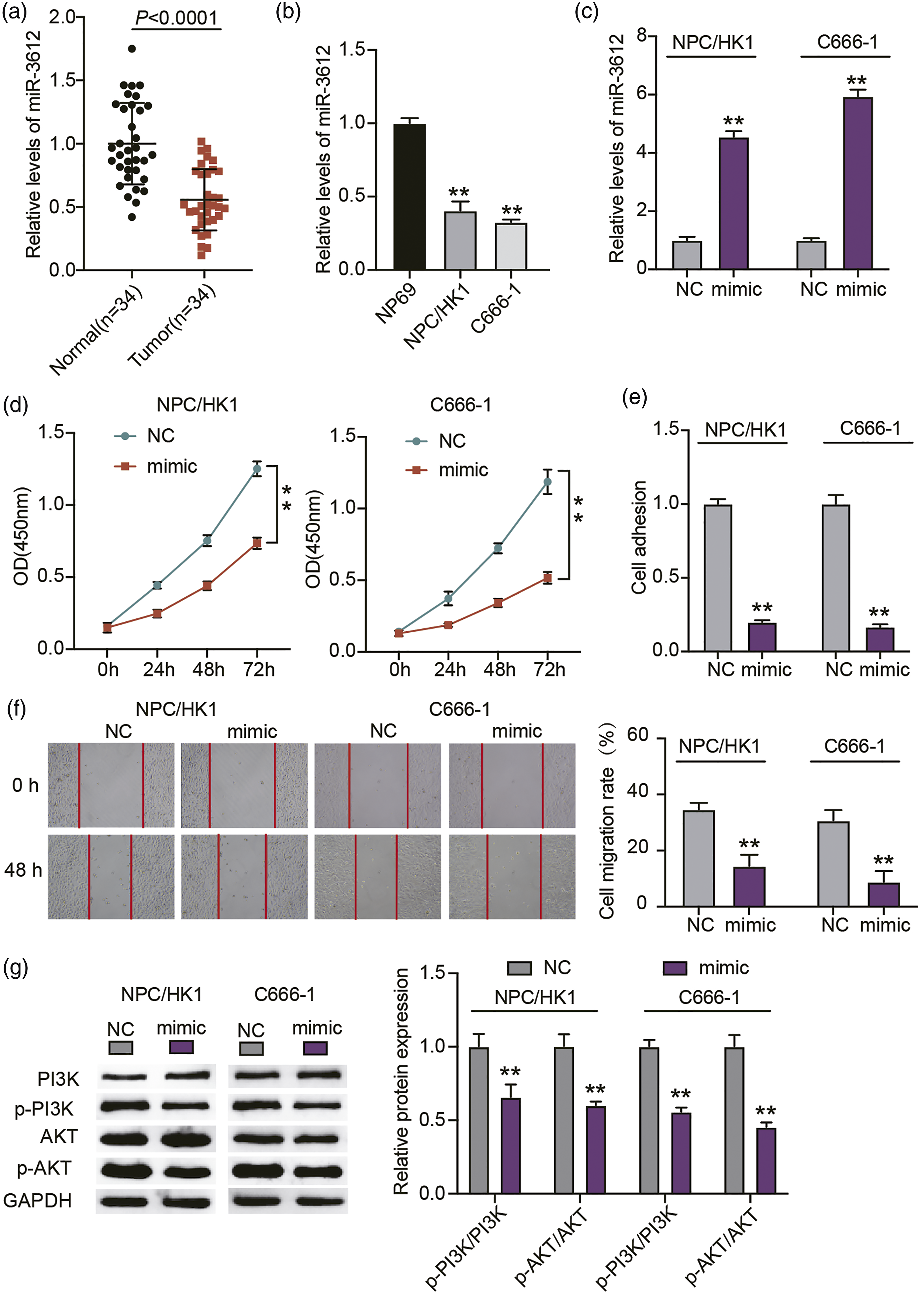
Overexpressing miR-3612 disables the tumorigenic potential of NPC in vivo
To assess how miR-3612 affects NPC progression in vivo, C666-1 cells stably infected with agomiR-3612 or agomiR-NC were administered into the nude mice to develop tumor xenografts. Figure 2(a) presents two representative photographs from the agomiR-3612 group and NC group. A decline in tumor volume can be observed in the agomiR-3612 group (Figure 2(b)). Furthermore, the transfection of agomiR-3612 considerably diminished the weights of the tumors (Figure 2(c)). Consistent with the outcomes in vitro, miR-361 overexpression also impeded NPC progression in vivo. Overexpressing miR-3612 disables NPC tumorigenic potential in vivo. 1 × 108 NPC/HK1 cells stably transfected with agomiR-3612 (10 μM) or agomirNC (10 μM) were administered into the mice. (a) Representative tumor photographs of NPC/HK1 xenografts bearing agomiR-3612 or agomirNC. (b) The volume of the tumors was recorded every four days after the establishment of the NPC tumor model. (c) At the 28th day, the mice were euthanized by means of CO2 inhalation prior the dissection and weighing of the tumor xenografts. **p < 0.001 vs. NC.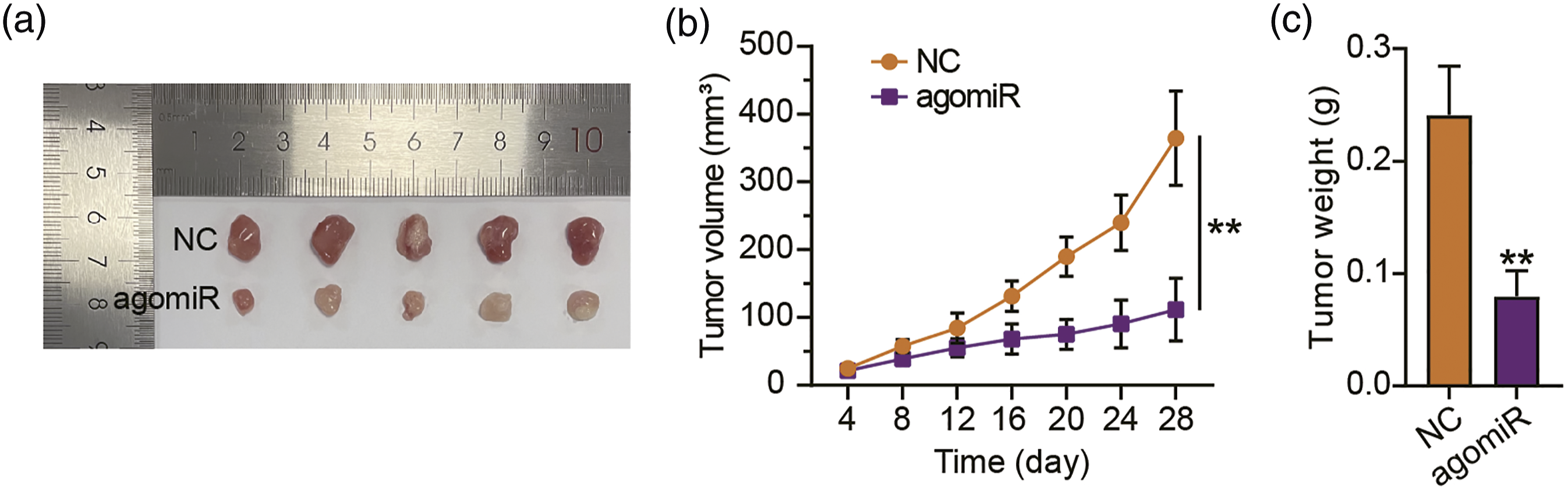
MiR-3612 targets THBS1 to regulate PI3K/AKT signaling pathway
MiRNA’s functionality is dependent on miRNA-mediated gene silencing.
16
Therefore, we employed TargetScan in our search for miR-3612 targets. Meanwhile, GSE12452 was used to screen the upregulated genes in NPC samples with adj. p < 0.05 and logFC ≥1. The results showed that 128 common genes from TargetScan and GSE12452 were screened out (Supplementary Figure 1A). After uploading 128 genes to STRING for KEGG enrichment, five genes (KITLG, THBS1, COL5A1, THBS2 and COL11A1) were found to be associated with PI3K-AKT-mTOR signaling pathway (Supplementary Figure 1B). The results of RT-qPCR identified that THBS1 was the most up-regulated gene in tumor tissues compared to other four genes (Supplementary Figure 1C–G). Therefore, THBS1 was selected as our interested gene. As shown in Figure 3(a), miR-3612 shared base pairing with the 3′UTR of THBS1. To investigate the interaction of miR-3612 with THBS1, we conducted luciferase reporter assays to quantify the luciferase activities mediated by the transfection of a combination of miR-3612 mimic/mimic NC and a THBS1-WT/MUT vector into the NPC/HK1 and C666-1 cells. As anticipated, a reduction in luciferase activity was detected upon the simultaneous transfection of THBS1-WT and miR-3612 mimic (Figure 3(b)). Meanwhile, no notable changes were detected among those with THBS1-MUT. A consistently poor expression of THBS1 was measured in NPC tissues (Figure 3(c)) as well as in NPC/HK1 and C666-1 cells (Figure 3(d)) when compared to the normal groups. An inverse correlation between miR-3612 and THBS1 further evidenced that miR-3612 targeted THBS1 (Figure 3(e)). After transfecting miR-3612 mimic or inhibitor into NPC/HK1 and C666-1 cells, THBS1 protein expression was reduced in miR-3612 mimic group, and enhanced in miR-3612 inhibitor group (Figure 4(a)). Moreover, miR-3612 mimic decreased the levels of p-PI3K and p-AKT, whereas miR-3612 inhibitor increased the levels of p-PI3K and p-AKT (Figure 4(b)). In summary, miR-3612 downregulates THBS1 expression and regulates PI3K/AKT signaling pathway. MiR-3612 targets THBS1. (a) The putative binding sequences for miR-3612 within the 3′UTR of THBS1 were determined via starBase analysis. (b) After co-transfection with the miR-3612 mimic or mimic NC, the relative 3′UTR-THBS1-WT/MUT-driven luciferase density of pMIR-Reporter luciferase reporters was determined in NPC/HK1 and C666-1 cells. **p < 0.001 vs. WT + NC, #p < 0.05 and ##p < 0.001 vs. Co-Mut + NC. (c) RT-qPCR quantification of THBS1 expression in NPC tumors and normal tissues. (d) RT-qPCR quantifying the THBS1 expression in NPC/HK1 and NPC cells (C666-1 and NP69). **p < 0.001 vs. NP69. (e) The correlation of miR-3612 expression with THBS1 expression in NPC tissues as analyzed via Pearson’s correlation coefficients. MiR-3612 regulates THBS1 expression and PI3K/AKT signaling pathway. MiR-3612 mimic (mimic), mimic-NC, miR-3612 inhibitor (inhibitor), and inhibitor-NC were individually delivered into the C666-1 and NPC/HK1 cells. After the 48 h transfection, (a) Western blotting, using anti-THBS1 antibodies, was conducted to assess THBS1 expression in the transfected NPC/HK1 and C666-1 cells. (b) Western blotting analysis (with antibodies against PI3K, p-PI3K, AKT, and p-AKT) of the critical effectors of the PI3K/AKT signaling pathway in transfected C666-1 and NPC/HK1 cells. **p < 0.001 vs. mimic-NC, #p < 0.05 and ##p < 0.001 vs. inhibitor-NC.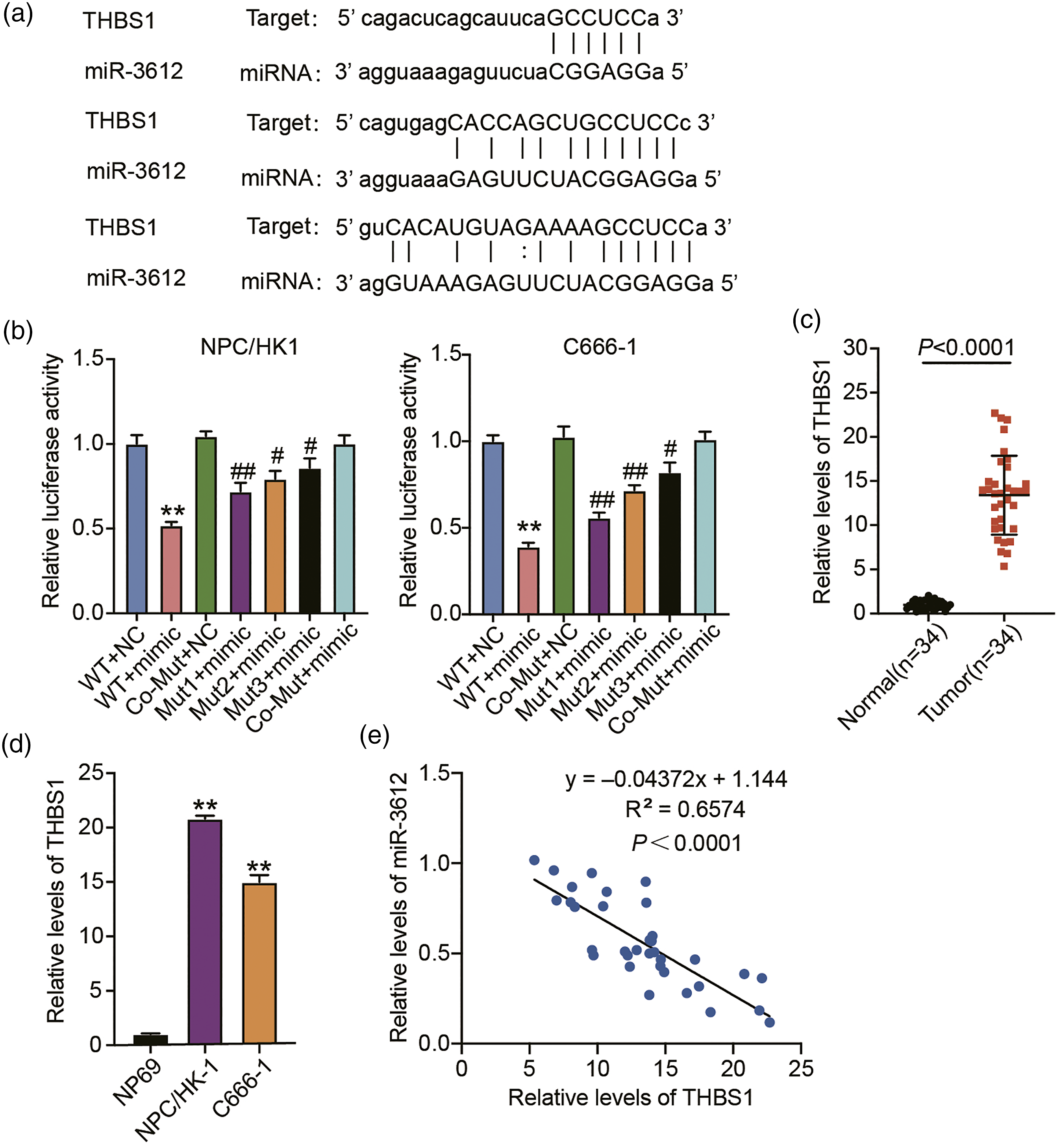

MiR-3612 overexpression reduces the oncogenic potential of THBS1 in NPC through the PI3K/AKT signaling pathway
To dissect THBS1’s critical role in the miR-3612-mediated NPC repression, we co-transfected miR-3612 mimic or NC with the THBS1 overexpressing vectors or NC into the NPC/HK1 and C666-1 cells. Western blotting analysis showed that miR-3612 mimic could successfully decrease the resultant THBS1 overexpression from the transfection of THBS1 overexpressing vectors (Figure 5(a)). Functionally, the NPC cell proliferation was enhanced in NPC/HK1 and C666-1 cells overexpressing THBS1. The boosted proliferation, however, was compromised upon the co-transfection of the miR-3612 mimic (Figure 5(b)). Furthermore, the enforced expression of THBS1 stimulated NPC cell migration, which was nullified by the miR-3612 mimic (Figure 5(c)). Consistently, the overexpression THBS1 increased cell adhesion, which was also offset by miR-3612 mimic transfection (Figure 5(d)). In addition, THBS1 overexpression in NPC cells increased the presence of p-AKT and p-PI3K. Nonetheless, this was also offset by the addition of the miR-3612 mimic (Figure 5(e)). To summarize, miR-3612 silences THBS1 expression and represses the THBS1-activated oncogenic PI3K/AKT signaling pathway, thereby retarding the progression of NPC. MiR-3612 overexpression reduces the oncogenic potential of THBS1 in NPC by regulating the PI3K/AKT signaling pathway. MiR-3612 mimic (mimic), mimic NC, THBS1 overexpressing vector (THBS1-OE), its empty vector (OE-NC), and miR-3612 mimic + THBS1-OE were individually delivered into the C666-1 and NPC/HK1 cells. After the 48-h transfection, (a) Western blotting, using anti-THBS1 antibodies, was conducted to assess THBS1 expression in the transfected NPC/HK1 and C666-1 cells. (b) The proliferative capacity of the transfected C666-1 and NPC/HK1 cells was evaluated via CCK-8 assays. (c) The migrative ability of the transfected C666-1 and NPC/HK1 cells was assessed by means of the scratch wound assays. (d) Tumor cell adhesion was assessed in the transfected C666-1 and NPC/HK1 cells. (e) Western blotting analysis (with antibodies against PI3K, p-PI3K, AKT, and p-AKT) of the critical effectors of the PI3K/AKT signaling pathway in transfected C666-1 and NPC/HK1 cells. **p < 0.001 vs. Empty vector, ##p < 0.001 vs. mimic-NC, &&p < 0.001 vs. OE + mimic.
Discussion
Here, miR-3612s anti-tumorigenic role in NPC in vitro and in vivo, has been revealed for the first time. The miR-3612-mediated repression of NPC was disrupted as THBS1 activated the PI3K/AKT signaling pathway. In brief, miR-3612 downregulated THBS1 to repress its pro-tumorigenic role in NPC through the inactivation of the PI3K/AKT signaling pathway.
Impairing tumorigenic potential is a common function for miR-3612 in different cancers.8,10,21,22 For example, miR-3612 targeting the NF receptor associated factor 4 led to the inactivation of NF-κB pathway, thereby suppressing esophageal squamous cell carcinoma. 9 Meanwhile, the elevated level of ETS transcription factor ELK3S effected by the sequestration of miR-3612 favors bladder cancer progression. 10 MiR-3612 strongly suppresses the malignant behaviors of esophageal squamous cell carcinoma cells. 22 Consistently, we detected a low miR-3612 expression in NPC cells and tissues. Upon miR-3612 overexpression, the NPC cells manifested a decrease in migration, adhesion, and proliferation and stimulated apoptosis. The in vivo results further evidenced miR-3612s tumor-suppressive function in NPC. Hence, our findings substantiate the anti-cancer role of miR-3612 in cancers.
To further examine the miR-3612-dependent regulation, we employed starBase and discovered that miR-3612 could recognize THBS1 3′UTR. This miRNA–mRNA interaction was verified by luciferase reporter assays. Hence, THBS1 captured our interest. THBS1 has been reported as a multifunctional extracellular matrix protein which performs subdivided roles in different tumors.11,23,24 An example of its complex function is the boost in tumor expansion and invasion induced by TGFβ1 as it stimulates THBS1 expression. 25 The highly expressed THBS1 impairs the activity of the vascular endothelial growth factor C, a well-known lymphangiogenic growth factor, which boosts colorectal cancerigenesis and progression. 26 Our data indicated an elevated expression of THBS1 in NPC cells and tissues. This overexpression of THBS1 led to an enhanced NPC cell migration, adhesion, and proliferation in vitro. Furthermore, Moradpoor et al. 27 reported that THBS1 was associated with the PI3K/AKT signaling pathway in breast cancer. The PI3K/AKT/mTOR pathway activation by THBS1 contributes to the malignant behaviors of human gastric cancer. 14 Therefore, we further unveiled THBS1’s action in PI3K/Akt signaling. Interestingly, we found high expressions of p-PI3K and p-AKT in NPC cells, following the overexpression of THBS1. Our findings provide further evidence for its oncogenic role in cancers. Moreover, we found that miR-3612 overexpression could nullify the pro-tumorigenic action of THBS1 in NPC cells. Therefore, miR-3612 downregulation and PI3K/AKT signaling pathway activation may be behind the pro-tumor action of THBS1 during NPC progression.
Some limitations in our work shall not be ignored. First, the miR-3612/THBS1/PI3K/AKT axis needs further verification in vivo. Furthermore, the PI3K/AKT inhibitor shall be studied to further confirm its role during miR-3612/THBS-mediated NPC progression. In addition, miR-3612 or THBS1 may each have different regulatory networks where they perform their actions, since they have different binding sites for their targets.
To conclude, we propose that miR-3612 inactivates the PI3K/AKT signaling pathway and represses NPC tumorigenesis by downregulating THBS1. Our work support that targeting miR-3612 may be a good candidate for therapeutic strategies against NPC.
Supplemental Material
Supplemental Material - MiR-3612 targeting THBS1 suppresses nasopharyngeal carcinoma progression by PI3K/AKT signaling pathway
Supplemental Material for MiR-3612 targeting THBS1 suppresses nasopharyngeal carcinoma progression by PI3K/AKT signaling pathway by Wei Zhang, Qiu Zhang, Qianbo Cui and Yu Xu in Human and Experimental Toxicology
Footnotes
Author contributions
WZ and QBC conducted the experiments and data analysis.
WZ and QZ conceived and designed the study. YX acquired the data.
QBC and YX performed the analysis and interpretation of data.
All authors have read and approved this manuscript.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by Disciplinary Fund of The Central Hospital of Wuhan [grant number 2021XK001].
Ethical approval
The Ethics Committee of Renmin Hospital of Wuhan University (Wuhan, China) approved this study. The processing of clinical tissue samples was done in strict compliance with the ethical standards of the Declaration of Helsinki. All patients signed a written informed consent. The animal experiment was conducted in accordance with the ARRIVE guidelines and was authorized by the Ethics Committee of Renmin Hospital of Wuhan University.
Informed consent
All patients signed a written informed consent. Consent for publication was obtained from all participants.
Data availability
All data generated or analyzed during this study are included in this article.
Supplemental Material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.