
Abstract
Intervertebral disc degeneration (IDD) is a complex degradative disorder associated with inflammation. Emodin, an anthraquinone derivative, possesses strong anti-inflammatory activity. This study focused on the in vitro therapeutic action of emodin in a cellular model of IDD. Human nucleus pulposus cells (NPCs) were stimulated with interleukin‐1β (IL-1β) to induce inflammation. Cell Counting Kit-8 and terminal deoxynucleotidyl transferase dUTP nick end labeling staining assays were performed to evaluate the viability and apoptosis of NPCs, respectively. Caspase-3 activity was measured to indirectly assess cell apoptosis. Western blot analysis was performed to detect protein expression levels. Reverse transcription‐polymerase chain reaction was performed for the detection of relative mRNA levels of tumor necrosis factor-α (TNF-α) and IL-6. Enzyme-linked immunosorbent assay was performed to analyze TNF-α and IL-6 secretion. Our results showed that emodin treatment mitigated IL-1β-induced reduction of cell viability in NPCs. Moreover, the increase in reactive oxygen species (ROS) production, apoptotic rate, and caspase-3 activity in IL-1β-stimulated NPCs was reduced by emodin treatment. Treatment with emodin also abolished IL-1β-induced inflammation in NPCs, as indicated by reduced secretion of IL-6 and TNF-α. Besides, the increase in expression levels of phosphorylated p65 and nuclear p65 in IL-1β-stimulated NPCs was suppressed by emodin treatment. Furthermore, inhibition of nuclear factor kappa B (NF-κB) activation with pyrrolidine dithiocarbamate aggravated the protective effects of emodin. These results suggested that emodin protected NPCs against IL-1β-induced apoptosis and inflammation via inhibiting ROS-mediated activation of NF-κB.
Introduction
Intervertebral disc degeneration (IDD) is a complex degradative musculoskeletal disorder, and remains to be a major cause of discogenic low back pain. 1 Patients with severe status of IDD have been shown to have poor quality of life with heavy economic burdens. Hence, it is crucial to develop novel therapeutic strategies to treat IDD or delay the process. A key characteristic of IDD pathology is disc degeneration, which is associated with inflammation, extracellular matrix (ECM) degradation, senescence, as well as cell loss (apoptosis). 2 The process of IDD includes a cascade of events, and among these, inflammatory response plays crucial roles in disease onset. 3 It has been reported that the degenerative processes first begins in the nucleus pulposus (NP). 4 NP cells (NPCs) are responsible for the maintenance of ECM, which is essential for the mechanical functionality of the intervertebral disc. In response to proinflammatory cytokines, ECM degradation and cell apoptosis occur in NPCs, which further induce inflammation. 5 Based on current knowledge of IDD pathogenesis, inflammation, ECM degradation, and apoptosis are considered as the hallmarks of IDD in NPCs.
Emodin (Figure 1) is an anthraquinone compound extracted from various pharmaceutical plants, such as Polygonum, Aloe vera, Rheum palmatum, and Giant knotweed.
6
Pharmacological studies have reported that emodin possesses a broad range of activities such as antioxidant, anti-cancer, anti-infection, anti-inflammatory, anti-diabetic, and immune regulation activities.
6
These beneficial effects of emodin provide evidence for its potential usage in preventing and treating related diseases. Interestingly, a previous study demonstrated that emodin induced the formation of osteoblasts and inhibited the activation and maturation of osteoclasts.
7
Moreover, emodin treatment suppressed inflammatory bowel disease-induced osteoporosis by inhibiting osteoclast formation.
8
These results imply that emodin promotes bone tissue regeneration and bone remodeling, indicating that emodin might serve as an effective agent for the prevention and treatment of metabolic bone disorders. However, the potential effects of emodin on IDD have not yet been reported. Chemical structure of emodin.
The inflammatory process, exacerbated by cytokines, like interleukin‐1β (IL-1β), is considered to be a key mediator of IDD. 9 Thus, we used IL-1β for the induction of inflammation in NPCs to generate an in vitro model of IDD. This study aimed to explore the therapeutic effects and underlying mechanism of action of emodin in IDD in vitro.
Materials and methods
Cell culture
Human NPCs (Cat. No: #4800; ScienCell Research Laboratories, Carlsbad, CA, USA) were cultured in NPC medium (Cat. No: #4801; ScienCell Research Laboratories) and grown in an incubator with 5% CO2 at 37°C.
Model establishment and experimental design
To induce inflammatory response, NPCs were treated with 10 ng/mL of IL-1β (Cat. No: 200-01B; Peprotech, Rocky Hill, NJ, USA), according to previously described protocols.10,11
To select the suitable concentration of emodin used in this study, NPCs were exposed to 0, 5, 10, 20, or 40 μM of emodin (Cat. No: HY-14,393; purity 99.39%; MedChemExpress, Monmouth Junction, NJ, USA) for 24 h. The treatment time was based on a previously used protocol. 10 Next, cells were treated with 5, 10, or 20 μM of emodin in the presence of 10 ng/mL of IL-1β.
To evaluate the effect of emodin, cells were grouped into control, emodin, IL-1β, IL-1β + emodin, and IL-1β + N-acetylcysteine (NAC) groups. In the control group, cells were treated with dimethyl sulfoxide (DMSO; Cat. No: D2660; Sigma-Aldrich, stokes Louis, MO, USA) for 24 h and the final concentration of DMSO was less than 0.1% (v/v). In the emodin group, cells were exposed to 20 μM of emodin for 24 h. In the IL-1β group, a cellular model was established using 10 ng/mL of IL-1β. In the IL-1β + emodin group, cells were exposed to 20 μM of emodin and 10 ng/mL of IL-1β for 24 h. In the IL-1β + NAC group, cells were exposed to 5 mM of NAC and 10 ng/mL of IL-1β for 24 h. NAC is a reactive oxygen species (ROS) scavenger, and its concentration used in this study was based on a previous paper. 12
To explore whether nuclear factor kappa B (NF-κB) signaling mediated the effects of emodin, 10 μM of pyrrolidine dithiocarbamate (PDTC; Cat. No: P8765; Sigma-Aldrich) was used to inhibit the activation of NF-κB as reported previously.13,14 Cells were grouped into control, IL-1β, IL-1β + emodin, IL-1β + PDTC, and IL-1β + emodin + PDTC groups. In the control, IL-1β, and IL-1β + emodin groups, cells were treated as described above. In the IL-1β + PDTC group, cells were incubated with 10 μM of PDTC and 10 ng/mL of IL-1β for 24 h. In the IL-1β + emodin + PDTC group, NPCs were exposed to 20 μM of emodin and 10 μM of PDTC in the presence of 10 ng/mL of IL-1β for 24 h.
Cell Counting Kit-8 assay
NPCs were adjusted to 5 × 104 cells/mL and then 100 μL of cell suspension was inoculated in a 96-well plate. After incubation for 24 h, cells were treated with 10 μL CCK-8 solution (Cat. No: C0040; Beyotime, Shanghai, China) to assess cell viability, according to the manufacturer’s protocol.
Measurement of ROS production
Intracellular ROS level in NPCs was detected using a fluorescent probe dichloro-dihydro-fluorescein diacetate (DCFH-DA; Cat. No: D6883; Sigma-Aldrich) as described previously. 15 Images were acquired using the Olympus BX61 fluorescence microscope (Olympus, Tokyo, Japan) and fluorescence intensity was analyzed using ImageJ software v1.8.0 (National Institutes of Health, Bethesda, MD, USA).
Terminal deoxynucle1otidyl transferase dUTP nick end labeling (TUNEL) staining
Apoptotic cell death was assessed by TUNEL staining as described previously. 16 Briefly, NPCs were harvested by trypsinization and then fixed with 4% paraformaldehyde (Cat. No: P0099; Beyotime) for 30 min. Next, cells were stained with TUNEL solution (Cat. No: C1086; Beyotime) for 1 h, followed by 4′,6-diamidino-2-phenylindole (DAPI; Cat. No: C1002; Beyotime) staining for 5 min. NPCs were then visualized under the Olympus BX61 fluorescence microscope (Olympus) and images were captured for the analysis of apoptotic rate.
Measurement of caspase-3 activity
To measure the enzyme activity of caspase-3, a caspase-3 activity assay kit (Cat. No: APT165; Sigma-Aldrich) was used. The absorbance was measured at a wavelength of 405 nm using a SpectraMax iD5 microplate reader (Molecular Devices, Sunnyvale, CA, USA).
Western blot analysis
Total protein and nuclear fractions were respectively extracted from NPCs using commercial kits (Cat. No: P0013B/P0027; Beyotime). The extracted proteins were then subjected to western blot analysis as described previously. 17 Antibodies against B-cell lymphoma 2 (Bcl-2; Cat. No: ab196495; 1:1000 dilution), Bcl-2-associated × protein (Bax; Cat. No: ab263897; 1:2000 dilution), p65 (Cat. No: ab16502; 1:2000 dilution), phosphorylated p65 (p-p65; Cat. No: ab194726; 1:500 dilution), lamin B (Cat. No: ab65986; 1:3000 dilution), caspase-3 (Cat. No: ab4051; 1:2000 dilution), cleaved caspase-3 (Cat. No: ab2302; 1:200 dilution), and β-actin (Cat. No: ab8227; 1:3000 dilution), as well as horseradish peroxidase-conjugated secondary antibody (Cat. No: ab6721; 1:10,000 dilution) were obtained from Abcam (Cambridge, MA, USA). Immunoreactive bands were visualized using ECL Plus Reagent (Cat. No: 32,134; Thermo Fisher Scientific, Waltham, MA, USA) and then semi-quantified with ImageJ software v1.8.0 (National Institutes of Health). β-Actin and lamin B acted as the internal reference genes.
Reverse transcription‐polymerase chain reaction (RT-PCR)
Total RNA was extracted from NPCs and transcribed into cDNA using a Reverse Transcription Kit (Cat. No: RR037A; Takara, Dalian, China). Subsequently, RT-PCR amplification was performed using SYBR Green PCR Master Mix (Cat. No: RR420L; Takara) as described previously. 18 The relative mRNA expression levels of tumor necrosis factor-α (TNF-α) and IL-6 were normalized to glyceraldehyde-3-phosphate dehydrogenase (GAPDH) and expressed as fold-changes. The following primer sequences were used: TNF-α, forward 5′-TCAGCCTCTTCTCCTTCCTG-3′ and reverse 5′-TTTGCTACAACATGGGCTACA-3′; IL-6, forward 5′-AGGAGACTTGCCTGGTGAAA-3′ and reverse 5′-GTCAGGGGTGGTTATTGCAT-3′; GAPDH, forward 5′-AATGGGCAGCCGTTAGGAAA-3′ and reverse 5′-GCGCCCAATACGACCAAATC-3′.
Enzyme-linked immunosorbent assay
The secretion levels of TNF-α and IL-6 in the culture supernatant of NPCs were quantified using corresponding ELISA kits (Cat. No: STA00D and SQ6000B, respectively) purchased from R&D Systems (Minneapolis, MN, USA), following the kit instructions.
Statistical analysis
Statistical analyses were performed using one-way analysis of variance (ANOVA) followed by Tukey’s post-hoc test with the GraphPad Prism v6.0 (GraphPad Software, San Diego, CA, USA). All quantitative data are expressed as mean ± standard deviation (SD). Values of p less than 0.05 were considered statistically significant.
Results
Emodin attenuated IL-1β-induced cell viability reduction and ROS production in NPCs
Treatment with emodin significantly decreased the viability of NPCs at a concentration of 40 μM (Figure 2(a)). Thus, for subsequent experiments, concentrations of 5, 10, and 20 μM were used. As shown in Figure 2(b), treatment with emodin attenuated the IL-1β-induced reduction in cell viability of NPCs in a dose-dependent manner. The concentration of 20 μM of emodin was selected for all following experiments. As shown in Figure 2(c) and (d), emodin or NAC treatment effectively inhibited IL-1β-induced ROS production in NPCs. Emodin attenuated cell viability reduction and ROS production in IL-1β-stimulated NPCs. (a) To evaluate the cytotoxicity of emodin, NPCs were exposed to 0, 5, 10, 20, or 40 μM of emodin for 24 h, followed by CCK-8 assay. (b) NPCs were exposed to 0, 5, 10, or 20 μM of emodin in the presence of 10 ng/mL of IL-1β for 24 h. CCK-8 assay was performed to evaluate cell viability. (c) NPCs were exposed to 20 μM of emodin or 5 mM of NAC in the presence of 10 ng/mL of IL-1β for 24 h. Representative images of ROS production are shown. (d) Quantification of relative ROS level in the different groups through analysis of fluorescence intensity. Three independent experiments were performed.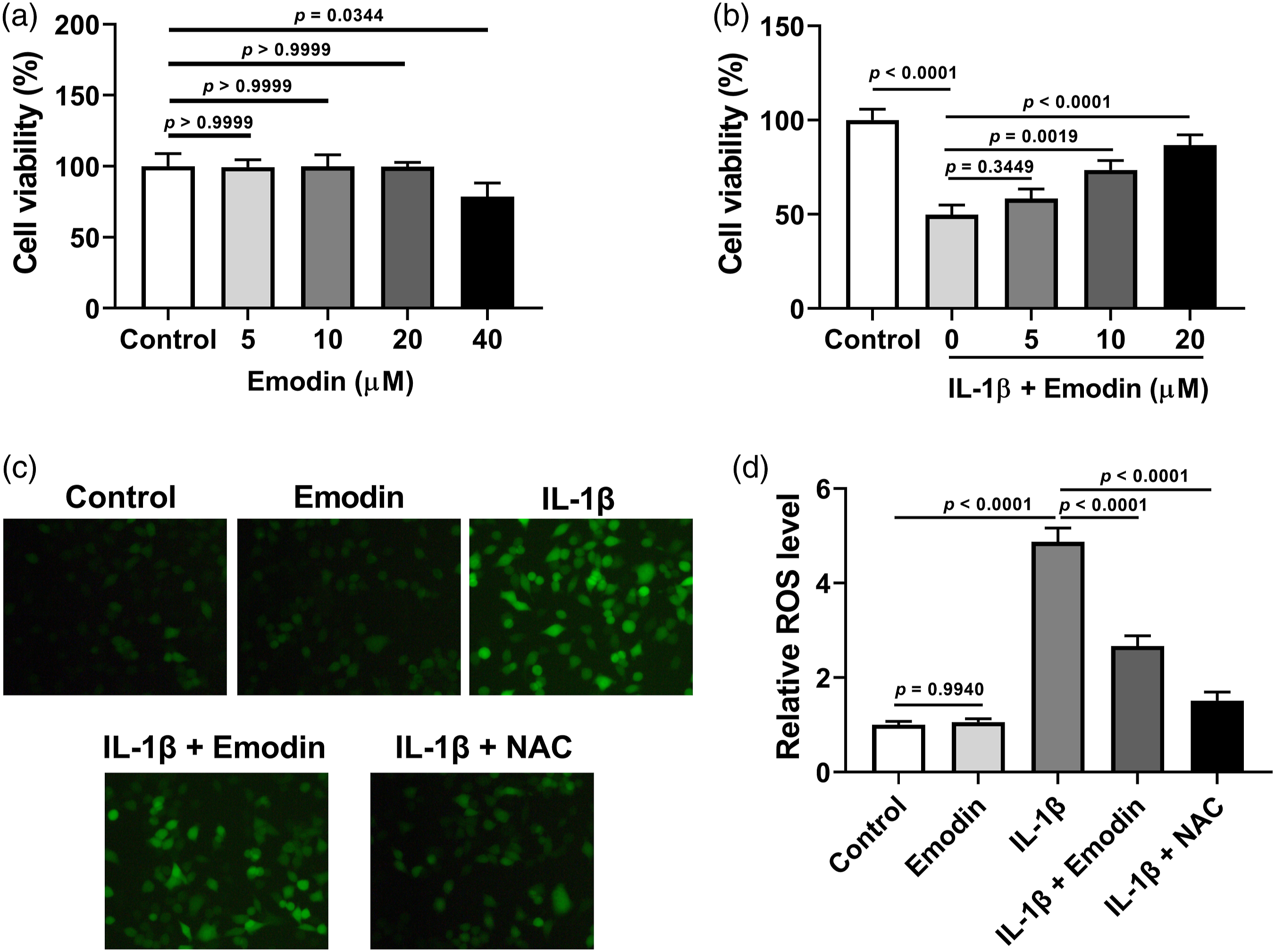
Emodin attenuated IL-1β-induced cell apoptosis in NPCs
Compared to the IL-1β treatment group, the emodin treatment group showed a significant decrease in the apoptotic rate of NPCs (Figure 3(a) and (b)). Treatment with emodin or NAC attenuated the IL-1β-induced increase in caspase-3 activity (Figure 3(c)). Additionally, there was a significant decrease in relative cleaved caspase-3/total caspase-3 and Bax/Bcl-2 levels in the IL-1β + emodin or NAC treatment group, as compared to the IL-1β treatment group (Figure 3(d)–(f)). Emodin attenuated IL-1β-induced cell apoptosis in NPCs. NPCs were exposed to 20 μM of emodin or 5 mM of NAC in the presence of 10 ng/mL of IL-1β for 24 h. (a and b) TUNEL staining assay was performed to detect apoptotic cell death in NPCs. (c) Quantification of relative caspase-3 activity. (d) Western blot analysis was performed to detect the expression levels of cleaved caspase-3, total caspase-3, Bcl-2, and Bax. (e) Quantification of relative cleaved caspase-3/total caspase-3 level. (f) Quantification of relative Bax/Bcl-2 level. Three independent experiments were performed.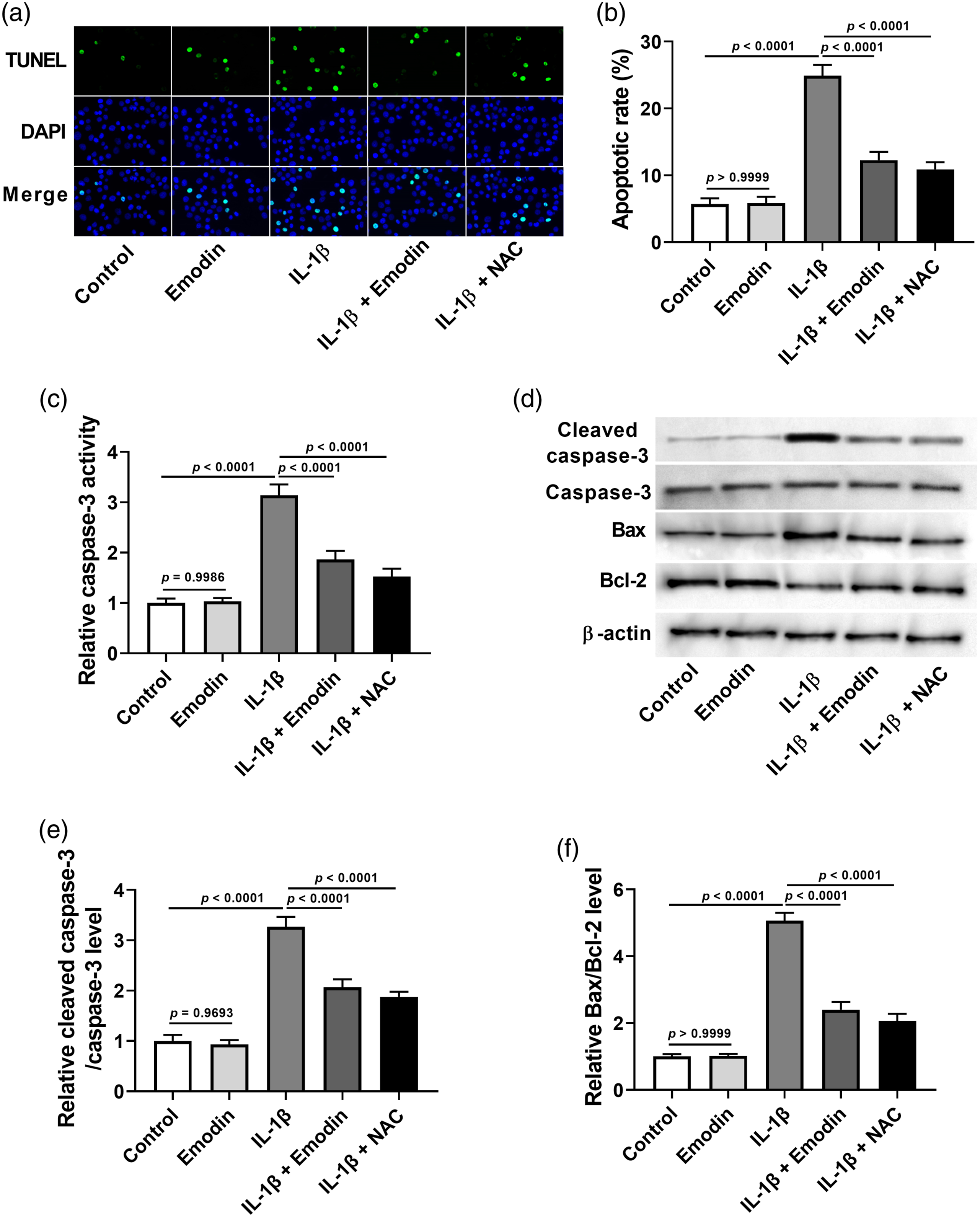
Emodin attenuated IL-1β-induced inflammation in NPCs
RT-PCR results demonstrated that the increased mRNA levels of TNF-α and IL-6 in IL-1β-induced NPCs were significantly downregulated by emodin or NAC treatment (Figure 4(a)–(d)). ELISA results further confirmed that treatment with emodin or NAC significantly decreased IL-1β-induced secretion of TNF-α and IL-6 in NPCs (Figure 4(g) and (h)). Emodin attenuated IL-1β-induced inflammation in NPCs. NPCs were exposed to 20 μM of emodin or 5 mM of NAC in the presence of 10 ng/mL of IL-1β for 24 h. (a and b) RT-PCR analysis for the determination of relative mRNA levels of TNF-α and IL-6. (c and d) ELISA was performed for evaluation of TNF-α and IL-6 secretion. Three independent experiments were performed.
Emodin attenuated IL-1β-induced activation of NF-κB in NPCs
Following stimulation with IL-1β, the relative p-p65/p65 level was dramatically increased in NPCs; however, treatment with emodin or NAC reduced the p-p65/p65 level (Figure 5(a) and (b)). Exposure of NPCs to IL-1β resulted in a significant increase in nuclear p65 expression, but emodin or NAC treatment suppressed this effect (Figure 5(c) and (d)). Emodin attenuated IL-1β-induced activation of NF-κB in NPCs. NPCs were exposed to 20 μM of emodin or 5 mM of NAC in the presence of 10 ng/mL of IL-1β for 24 h. (a) Western blot analysis was performed to detect the expression levels of p65 and p-p65. (b) Quantification of relative p-p65/p65 level. (c) Western blot analysis was performed to detect the expression levels of nuclear p65. (d) Quantification of relative nuclear p65 level. Three independent experiments were performed.
Inhibition of NF-κB aggravated the effects of emodin on IL-1β-treated NPCs
NPCs treated with emodin or PDTC displayed decreased apoptotic rates as compared to NPCs treated with IL-1β. Moreover, treatment with emodin + PDTC in the presence of IL-1β enhanced the decrease in apoptotic rate as compared to treatment with IL-1β + emodin (Figure 6(a) and (b)). The inhibitory effect of emodin on IL-1β-induced caspase-3 activity was enhanced by treatment with PDTC (Figure 6(c)). Treatment with emodin or PDTC inhibited the increase of cleaved caspase-3/total caspase-3 and Bax/Bcl-2 levels. The relative cleaved caspase-3/total caspase-3 and Bax/Bcl-2 levels were further decreased in the IL-1β + emodin + PDTC treatment group compared to those in the IL-1β + emodin treatment group (Figure 6(d)–(f)). Furthermore, treatment with emodin or PDTC inhibited the increase in TNF-α and IL-6 mRNA levels (Figure 7(a) and (b)) and secretion (Figure 7(c) and (d)). However, PDTC treatment enhanced the inhibitory effect of emodin on IL-1β-induced increase in TNF-α and IL-6 mRNA levels (Figure 7(a) and (b)) and secretion (Figure 7(c) and (d)). Inhibition of NF-κB aggravated the effect of emodin on IL-1β-induced apoptosis. NPCs were exposed to 20 μM of emodin and/or 10 μM of PDTC in the presence of 10 ng/mL of IL-1β for 24 h. (a and b) Apoptotic rate was evaluated by TUNEL assay. (c) Quantification of relative caspase-3 activity. (d) Western blot analysis was performed to detect the expression levels of cleaved caspase-3, total caspase-3, Bcl-2, and Bax. (e) Quantification of relative cleaved caspase-3/total caspase-3 level. (f) Quantification of relative Bax/Bcl-2 level. Three independent experiments were performed. Inhibition of NF-κB aggravated the effect of emodin on IL-1β-induced inflammation. NPCs were exposed to 20 μM of emodin and/or 10 μM of PDTC in the presence of 10 ng/mL of IL-1β for 24 h. (a and b) RT-PCR analysis for the determination of relative mRNA levels of TNF-α and IL-6. (c and d) ELISA was performed for evaluation of TNF-α and IL-6 secretion. Three independent experiments were performed.

Discussion
As an important member of the IL-1 family, IL-1β has gained much attention in recent years due to its strong pro-inflammatory activity in inflammation-related diseases. 19 A previous study demonstrated a significant increase in IL-1β level in degenerative intervertebral discs. 20 IL-1β has been shown to mediate multiple pathological processes in IDD progression, such as inflammatory response, cell proliferation, oxidative stress, cellular senescence, apoptosis, and ECM degradation. 21 Recently, increasing research has revealed the link between elevated IL-1β level and oxidative stress in IDD onset and development. 22 IL-1β was found to dramatically increase the production of intracellular ROS, which can induce oxidative stress. 23 ROS-mediated oxidative stress not only reinforces inflammatory response and ECM degradation, but also promotes functional cell loss in the microenvironment of intervertebral discs, leading to the progression of IDD. 24
It has been reported that emodin inhibits oxidative liver injury through preventing ROS production and mitochondrial dysfunction via the adenosine monophosphate-activated protein kinase signaling pathway. 25 Emodin protects human neuroblastoma SH-SY5Y cells against fluoride-induced synaptic impairment and ROS-mediated oxidative stress by modulating the extracellular signal‐regulated kinases 1/2/nuclear factor erythroid 2-related factor 2 (Nrf2)/heme oxygenase-1 pathway. 26 Besides, emodin mitigates cisplatin-induced augmentation of ROS and oxidative stress in osteosarcoma MG63 cells through activation of the Nrf2/antioxidant response element signaling pathway. 27 In a previous study by Song et al., 28 data suggested that emodin inhibited oxidized fish oil-induced ROS generation in the liver of teleost Megalobrama amblycephala. Moreover, emodin reduced mitochondrial-dependent apoptosis, repressed mitochondrial ROS generation, and accelerated adenosine triphosphate recovery both in a mouse model of renal ischemia/reperfusion injury and in a cellular model of hypoxia/reoxygenation injury. 29 Emodin has also been reported to exert anti-inflammatory effects in various pathological conditions. Emodin attenuated silica-induced inflammation and lung tissue fibrosis through inhibiting epithelial-mesenchymal transition, apoptosis, and inflammation. 30 Moreover, emodin suppressed lipopolysaccharide (LPS)-induced pulmonary inflammatory injury in rats via regulating the mammalian target of rapamycin/hypoxia-inducible factor-1α/vascular endothelial growth factor signaling pathway. 31 Emodin also suppressed chronic intestinal inflammation associated with carcinogenesis through inhibiting the recruitment of inflammatory cells and expression of cytokines and pro-inflammatory enzymes in the tumor microenvironment. 32 Furthermore, emodin was shown to inhibit the LPS-induced production of inflammatory cytokines, including TNF-α, IL-1β, and IL-6, by activating autophagy in RAW 264.7 cells. 33 Sun et al. 34 showed that emodin alleviated high glucose-induced oxidative stress, inflammation, and ECM accumulation in mesangial cells. Besides, recent studies have suggesetd that emodin exerted anti-inflammatory effects in certain murine models of inflammatory disease.35,36 Here, our results also demonstrated that emodin treatment abolished the increase in IL-1β-induced expression of IL-6 and TNF-α in NPCs. These data suggest that emodin protects NPCs against IL-1β-induced ROS production, cell apoptosis, and inflammation.
NF-κB, a redox-sensitive nuclear transcription factor, is closely associated with a diverse range of biological processes through regulating the expression of target genes. 37 Activated NF-κB translocates to the nucleus, where it induces the activation of a variety of inflammation-related genes, such as IL-6 and TNF-α.38–41 A regulatory relationship between ROS and NF-κB signaling has been demonstrated. 42 ROS regulates NF-κB activation through promotion of nuclear translocation of NF-κB.43,44 With the progression of IDD, ROS levels in the intervertebral discs increase, which further activates the NF-κB pathway and promotes the release of inflammatory factors, ultimately leading to the promotion of NP degeneration. 45 Feng et al. 46 reported that the NF-κB pathway was a downstream signaling pathway of ROS in NPCs, which participated in high oxygen tension-induced premature senescence of NPCs. Jiang et al. showed that resveratrol alleviated mechanical overloading-induced senescence of NPCs via regulating the ROS/NF-κB pathway. 47 Yao et al. 48 suggested that marein protected NPCs against high glucose-induced injury and ECM degradation partly through inhibiting the ROS/NF-κB pathway. Recently, naringin was reported to attenuate disc degeneration in a rat model of IDD by inhibiting the ROS/NF-κB pathway. 49 Moreover, the suppressive effects of osteogenic protein-1, 17β-estradiol, and N-cadherin on NPC senescence were shown to be involved in the ROS/NF-κB pathway.50–52 In this study, our results showed that treatment with emodin or NAC inhibited IL-1β-induced ROS generation and NF-κB activation. Moreover, inhibition of NF-κB activation by PDTC enhanced the protective effects of emodin on IL-1β-induced cell apoptosis and inflammation in NPCs. These data suggest that emodin exerts its effects via inhibiting the ROS-mediated activation of NF-κB.
However, this study had certain limitations. Firstly, our results only confirmed the suppressive effect of emodin on ROS-mediated NF-κB activation, while other potential underlying mechanisms were not studied. Potential upstream factors of the NF-κB pathway, such as sirtuin 1, visfatin, or myeloid differentiation factor 88, as reported in previous studies,35,36,53 should also be explored in the future. Furthermore, in vitro studies are different from in vivo. Hence, to better understand the role of emodin in vivo, animal models of IDD should be established and used for further research.
In conclusion, emodin protected NPCs against IL-1β-induced cellular injury via inhibiting ROS-mediated activation of NF-κB (Figure 8). These findings suggest that emodin might serve as a promising therapeutic agent in the prevention and treatment of IDD.
54
A schematic diagram depicting that emodin protected NPCs from IL-1β-induced cellular injury via inhibiting ROS-mediated activation of NF-κB.
Footnotes
Data availability statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Declaration of conflict of interest
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.