
Abstract
Acrylamide (AA) at high exposure levels is neurotoxic, induces testicular toxicity, and increases dominant lethal mutations in rats. RNA-sequencing in testes was used to identify differentially expressed genes (DEG), explore AA-induced pathway perturbations that could contribute to AA-induced testicular toxicity and then used to derive a benchmark dose (BMD). Male F344/DuCrl rats were administered 0.0, 0.5, 1.5, 3.0, 6.0, or 12.0 mg AA/kg bw/d in drinking water for 5, 15, or 31 days. The experimental design used exposure levels that spanned and exceeded the exposure levels used in the rat dominant lethal, 2-generation reproductive toxicology, and cancer bioassays. The time of sample collection was based on previous studies that developed gene expression–based BMD. At 12.0 mg/kg, there were 38, 33, and 65 DEG (P value <.005; fold change >1.5) in the testes after 5, 15, or 31 days of exposure, respectively. At 31 days, there was a dose-dependent increase in the number of DEG, and at 12.0 mg/kg/d the top three functional clusters affected by AA exposure were actin filament organization, response to calcium ion, and regulation of cell proliferation. The BMD lower 95% confidence limit using DEG ranged from 1.8 to 6.8 mg/kg compared to a no-observed-adverse-effect-level of 2.0 mg/kg/d for male reproductive toxicity. These results are consistent with the known effects of AA on calcium signaling and cytoskeletal actin filaments leading to neurotoxicity and suggest that AA can cause rat dominant lethal mutations by these same mechanisms leading to impaired chromosome segregation during cell division.
Keywords
Introduction
Acrylamide (AA) is a well-established human and rodent neurotoxin and a widely used industrial commodity chemical in polymer manufacturing. It is also regarded as a probable human carcinogen (International Agency for Research on Cancer), 1 and in rats, AA affects male reproductive performance and induces dominant lethal mutations. 2 Renewed interest in the toxicology of AA was sparked by the demonstration that AA is found at low levels in a variety of common foods and thus poses a hazard for widespread human exposure; elevated AA levels in food are related to carbohydrate content and cooking temperature. 3 The European Food Safety Authority (EFSA) has reviewed the available data on the toxicology of AA in food and has published its first full-risk assessment of AA in food. 4 The EFSA concluded that the current levels of dietary exposure to AA are a potential health concern, and efforts should be focused on reducing the levels of AA in the diet.
AA is considered a prototypic neurotoxicant that causes distal axonopathy characterized by degeneration of nerve terminals. 5 Its binding to neurofilament and microtubule proteins obtained from rat spinal cord and brain was demonstrated decades previously. 6 In addition, AA exposure affects neuronal calcium and calmodulin-dependent kinases that mediate the phosphorylation of neurofilament proteins, critical components of the neuronal cytoskeleton, and are cellular targets associated with neural degenerative diseases. 7,8 More recent proteomic studies demonstrate that AA is a weak electrophile that binds to cysteine thiolate groups of presynaptic proteins and proteins involved in axonal transport leading to aberrant nerve terminal processes at high AA exposures, producing a characteristic axonal neuropathy in humans and rodents. 5 The binding of AA to neurofilaments and motor proteins involved in axonal transport is speculated as the basis not only for AA neurotoxicity 9 but also for the genotoxic effects of AA. 10 This led us to question whether the two toxicities associated with AA exposure are mechanistically related.
The genotoxicity of AA has been reviewed by a number of authors and was part of the EFSA assessment of AA. 4,11,12 We have recently shown that AA exposure to rats at 12.0 mg/kg body weight (bw) for 30 days does not result in a biologically significant increase in either Pig-a gene mutations or micronuclei; in mice, at exposure levels of up to 24.0 mg/kg/d for 30 days, an increase in MN but not in Pig-a gene mutation was observed. 13 These studies indicate that AA is not directly genotoxic in rat bone marrow and that genotoxicity in mice is primarily associated with chromosomal alterations but not point mutations. Although several studies suggest that the genotoxic effects of AA specifically in mice occur via metabolism by CYP2E1 to glycidamide (GLY), there is limited evidence to support a causal relationship in rats for GLY-induced genotoxicity. 14 -16
The dominant lethal effects of AA in rats are of paternal origin. 12 Subchronic and short-term high-dose exposures of male rats to AA induces a spectrum of testicular toxicity including degeneration of seminiferous tubules, Sertoli and germ cell degeneration, formation of multinucleated giant and apoptotic cells in seminiferous tubules, decreased Leydig cell viability, aberrant sperm morphology, decreased sperm count, and motility. 1,17,18 The formation of multinucleated cells is hypothesized to be due to incomplete meiosis impairing spermatogenesis, suggesting an impact of AA on cellular cytoskeleton. AA-induced germ cell genotoxicity in rats is believed to result indirectly from its ability to form adducts with proteins rather than DNA. AA is proposed to interact with kinesin motor proteins resulting in compromised chromosomal segregation during the cell division (eg, giant cells) or through direct alkylation of sperm protamines. 10,19 AA administration in rats also induces an increase in biomarkers associated with oxidative stress including an increase in malondialdehye, decreases in glutathione (GSH) levels, and GSH peroxidases. 20
Since AA exposure is also known to affect calcium signaling in rat neurons, there is cause to suspect that AA can also affect calcium homeostasis in testes since AA is well distributed throughout the rat including testes. 12 Ca2+ is critical in different cellular signaling processes and has been long recognized as an intracellular messenger. 21 Ca2+ concentration must be finely regulated both intra- and extracellularly and Ca2+ concentration is maintained within strict limits by a variety of mechanisms including calcium channel pumps and intracellular cellular compartmentalization. 21 Calcium signaling plays a key role in spermatogenesis, from intracellular signaling within Sertoli cells in response to testosterone, to maturation of spermatozoa and sperm motility, known cellular targets for AA toxicity in testes. 22
These studies taken together indicate that the testicular toxicity of AA in rats that can result in dominant lethal mutations may involve multiple mechanisms including interaction of AA with cytoskeletal proteins and motor proteins involved in chromosomal segregation, oxidative stress, and impact on calcium signaling all known intracellular targets for AA neurotoxicity. 6,9,23 -25 Transcriptional profiling of target tissues following chemical exposure is a highly informative means to provide an unbiased assessment of mode of action (MOA) and can be used for quantitative estimation of toxicity. 26 -31 One previous study using microarray-based profiling at a single high dose (60 mg/kg for 5 days) confirmed many of the pathological changes observed in testes including an induction of apoptotic genes, genes specific to testicular function (eg, dynein-associated protein RKM23), and induction of specific CYP450 messenger RNAs (mRNAs); at this high dose of AA, there was no indication of a p53 DNA damage response commonly observed for genotoxic compounds. 17 Since gene expression changes are time- and dose dependent, this assessment of AA testicular toxicity by expression profiling using a single high dose is not sufficient for use in a risk assessment. In this study, we used next-generation RNA-sequencing (RNA-seq) to measure transcriptional changes occurring over time in the testes of male rats following subchronic oral exposure to a wide range of AA doses. Our goals were (i) to examine the temporal concordance and dose–response of the transcriptional data in support of an MOA for AA testicular toxicity in rats, (ii) to perform dose–response modeling and a benchmark dose (BMD) for testicular toxicity in rats, and (iii) to derive a toxicogenomics-based point of departure for potential use in human health risk assessment of AA.
Methods
Animal Exposures
Male F344/DuCrl rats were purchased from Charles River Laboratories (Raleigh, NC) and acclimatized for 12 days prior to the onset of the experiments, at which point animals were 8 weeks of age. The animal use protocol for this study was approved by the Integrated Laboratory Systems Inc Institutional Animal Care and Use Committee. All procedures were carried out in an Association for Assessment and Accreditation of Laboratory Animal Care (AAALAC)-accredited facility (AAALAC International File number: 000810). Animals were on 12/12-hour light/dark cycle (lights on:off, 0800:2000). Animals were housed in polycarbonate cages and fed NIH-07 Irradiated (Harlan Teklads Diet, Madison, Wisconsin) ad libitum. The animals were assigned to a dose group by bw, such that mean bw of each group was not statistically different from any other group using analysis of variance (Statistical Analysis System version 9.2, SAS Institute, Cary, North Carolina). AA (CAS no 79-06-1; Sigma-Aldrich, St. Louis, Missouri; batch BCBH1775V) was provided in drinking water by reverse osmosis treated tap water (City of Durham, North Carolina) ad libitum. AA was prepared weekly in drinking water, and concentrations were adjusted based upon the previous week’s group mean bw and water consumption data.
Rats (6 males per exposure group) were administered 0.0, 0.5, 1.5, 3.0, 6.0, or 12.0 mg AA/kg bw/d in drinking water (Figure 1) adjusted weekly based on animal weights. On days 5, 15, or 31, animals were stratified in order, 1 animal from each dose group throughout the dose groups then repeated, and euthanized by carbon dioxide asphyxiation with death was confirmed by exsanguination. Testes were weighed to the nearest 0.0001 g.
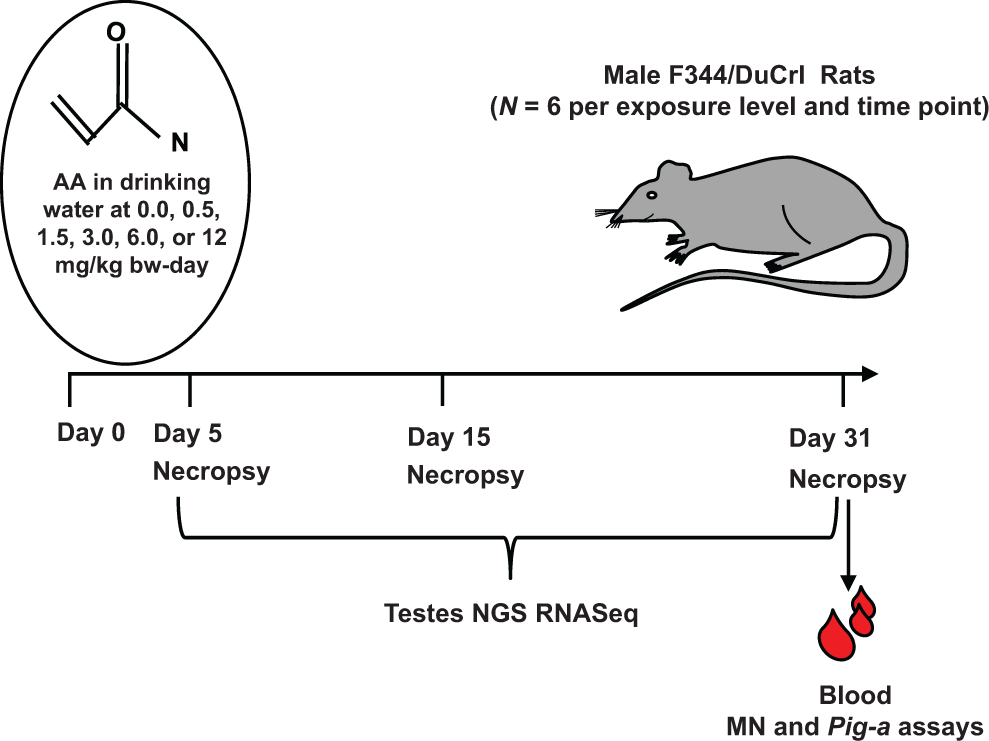
Experimental design for rat testes dose- and time-dependent toxicogenomics.
Expression Profiling by Next-Generation Sequencing of RNA: RNA-Seq
Frozen testes samples were homogenized in 2-minute intervals using 7.0 mm stainless steel beads and a Retsch Mixer Mill MM 400 (Verder Scientific, Newtown, Pennsylvania) at 20 Hz. Frozen samples were homogenized for 30 seconds in TRIzol® reagent using a PowerGen 125 at 15 000 to 20 000 rpm, until no tissue was visible, indicating complete homogenization. For the testes samples, total RNA was isolated from homogenized samples using TRIzol® reagent according to the manufacturer’s protocol (Invitrogen, Waltham, Massachusetts). RNA quantity and A260/280 ratios were measured using a Nanodrop ND-1000 spectrophotometer (Thermo Fisher Scientific, Waltham, Massachusetts). RNA quality was assessed using an Agilent RNA 6000 Nano kit and an Agilent 2100 Bioanalyzer according to the manufacturer’s protocol (Agilent Technologies, Santa Clara, California). The RNA integrity numbers (RINs) were generated using the Agilent 2100 Expert Software (Version. B.02.08.SI648 Agilent Technologies). Only samples with RINs >7.0 were used for mRNA analysis.
Poly-A RNA enrichment was performed for each sample using 1.5 µg of total RNA according to the New England BioLabs Ultra Directional RNA library prep for Illumina kit (New England Biolabs Inc, Ipswich, Massachusetts). The number of biological replicates was 6 per each of 6 dose group (including controls), per time point, that is N = 6 animals/dose × 6 doses × 3 time points = 108 samples in total. A dual barcode scheme was used to multiplex 36 samples together for each RNA-Seq experiment; for each individual time point, the control and treated rats (n = 36) were run together on a flow cell using an Illumina NextSeq500 (San Diego, California). RNA-Seq was done to approximately 10 million reads per sample.
Bioinformatics to Identify Differentially Expressed Genes and Gene Ontology Pathway Analysis
A number of quality control metrics were employed to assess the quality of RNA-Seq data generated for each sample. This included per base quality, per base sequence content, and the presence of adaptors. The RNA-Seq reads were aligned with TopHat (https://ccb.jhu.edu/software/tophat/index.shtml) using the rat rn6 genome using (rat rn6: http://useast.ensembl.org/Rattus_norvegicus/Info/Index). RNA-Seq data were normalized using Cuffnorm to the expression levels from the different RNA-Seq libraries (http://cole-trapnell-lab.github.io/cufflinks/cuffnorm/index.html). Cufflinks was used to conduct to identify differentially expressed genes (DEG; http://cole-trapnell-lab.github.io/cufflinks/). Principal component analysis was used to visualize patterns in the data, and hierarchical clustering was used to check for any groups in the data. For each treatment day (days 5, 15, or 31), the RNA-Seq expression signal was compared from each exposure level (0.5, 1.5, 3.0, 6.0, and 12.0) to the corresponding unexposed control from the same day.
The DEG with P value ≤.005 and fold change ≥±1.5 were uploaded into the “Functional Annotation Tool” of the Database for Annotation, Visualization and Integrated Discovery (DAVID), available at https://david.ncifcrf.gov/, to determine the most enriched gene ontology terms, using ENSEMBLE IDs of the identified DEGs, False discovery rate-adjusted Fisher exact P value <.05, and DAVID’s default settings.
Reverse Transcription
Reverse transcription was performed using the High-Capacity cDNA Reverse Transcription Kit (Applied Biosystems™ [ABI], Foster City, California) according to the manufacturer’s protocol for reaction setup and thermal cycling conditions (25°C for 10 minutes, 37°C for 120 minutes, 85°C for 5 minutes, and 4°C hold). The cDNA was then diluted in RNAse-free water to 20 ng/µL.
Quantitative Real-Time Polymerase Chain Reaction
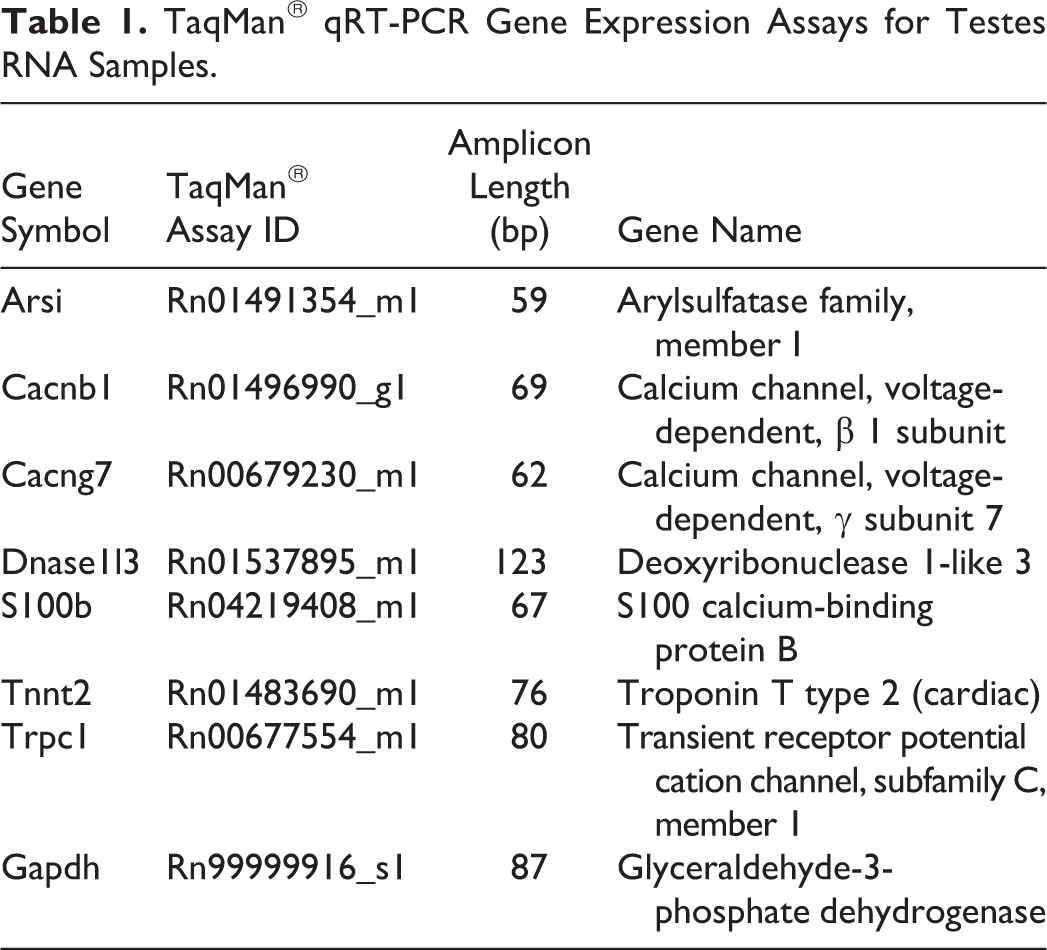
The real-time polymerase chain reaction (qRT-PCR) reactions were amplified using 20 ng of cDNA template, TaqMan® Fast Advanced Master Mix (ABI), and TaqMan® Gene Expression Assays (ABI) as indicated in Table 1. The qRT-PCR was performed using a ViiA™ 7 qRT-PCR System (ABI) with manufacturer’s recommended thermal cycling conditions for TaqMan® Fast Advanced Master Mix (50°C for 2 minutes, 95°C for 20 seconds, 95°C for 1 second, and 60°C for 20 seconds for 40 cycles). The mRNA levels were quantified using the cycle threshold (CT) values for the target genes of interest and the endogenous reference gene, Gapdh. Data were reported as the relative quantification (RQ; where RQ = 2–(ΔΔCT)) of treated animals normalized to the control group using DataAssist™ v3.0.1 software (ABI).
TaqMan® qRT-PCR Gene Expression Assays for Testes RNA Samples.
Quantitative RT-PCR Statistical Analysis
ΔCTs were analyzed using Analyse-It® v2.30 (Analyse-It Software, Leeds, UK) software with a 95% confidence interval for the vehicle control group.
Bench Mark Dose Modeling of Gene Expression
The BMD modeling of gene expression data 32 was performed, as described elsewhere, 33 using the expression values of all DEG identified on day 31, followed by data examination in the BMDExpress Data Viewer. 34
Results
RNA-Seq Quality Metrics, Principal Component Analysis, and Hierarchical Clustering
Quality metrics for each sequence read indicated that all samples were of good quality. Sequence alignment by tophat indicated that 93% of the sequenced reads aligned to the rat rn6 genome/transcriptome with 60% to 70% of the aligned reads from exons. The percentages of aligned reads were within the expected range. Principal component analysis indicated a few outliers for day 5 and day 15, with no obvious outliers observed for day 31. After removal of outliers from day 5 and day 15, a dose-dependent grouping of samples was observed for each time point by hierarchical clustering.
Differentially Expressed Genes (DEG)
The number of DEG (1.5-fold, P < .005) compared to the vehicle controls was examined with dose and for each day (days 5, 15, and 31). Overall, a small number of transcripts were detected as differentially expressed on days 5, 15, and 31. The largest number of DEG (65 transcripts) was observed at the highest AA exposure level (12.0 mg/kg/d) and for the longest duration of AA exposure (day 31; Figure 2). At 6.0 and 12.0 mg/kg, Cyp2a1 rat testosterone 7 α-hydroxylase was increased in a dose-dependent manner: 6.0 mg/kg/d (1.5-fold) and 12.0 mg/kg/d (2.0-fold). The list of the top 10 DEG across all exposure levels and time points is in Supplemental Table 1.
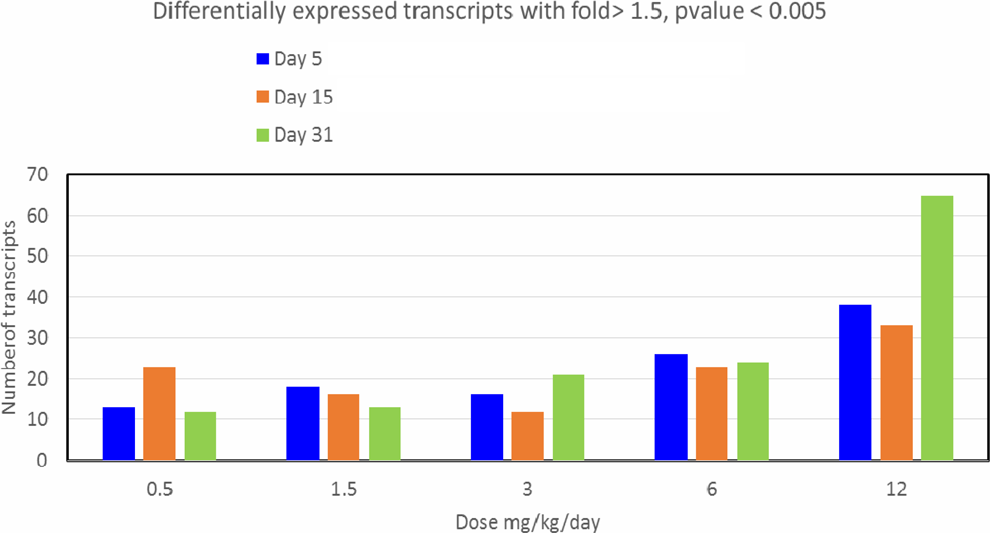
Number of differentially expressed genes with dose and time in testes of acrylamide (AA)-exposed rats.
Gene Ontology and Pathway Enrichment Analysis
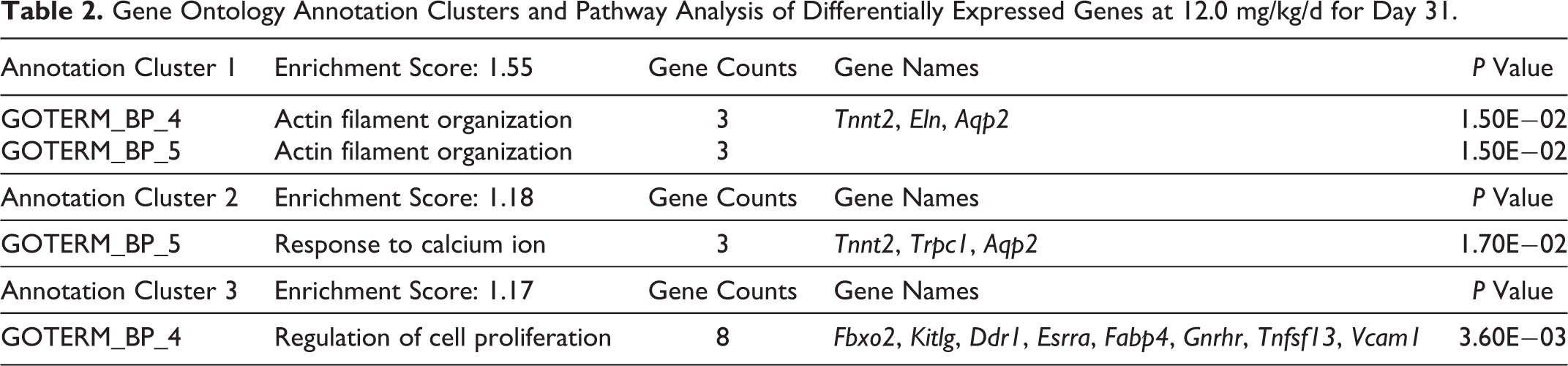
The list of DEG compared to the vehicle controls was examined with exposure level and for each day (days 5, 15, and 31) using gene ontology and genomic pathway analysis features of DAVID. The DEG for day 31 showed a dose–response, and 12.0 mg/kg showed the highest number of DEG. The top 3 gene ontology, pathway enrichment analyses, and DEG for each pathway for day 31 at 12.0 mg/kg is shown in Table 2. The top 3 annotation clusters for AA exposed rats on day 31 at 12.0 mg/kg were actin filament organization, calcium ion signaling, and cell proliferation. The list of the top 3 gene ontology pathways and the pathway enrichment across all exposure levels and time points is in Supplemental Table 2.
Gene Ontology Annotation Clusters and Pathway Analysis of Differentially Expressed Genes at 12.0 mg/kg/d for Day 31.
qRT-PCR of Target Genes Identified by NGS RNA-Seq
To confirm the RNA-Seq DEG for proposed calcium ion signaling-based MOA in testes and use of this pathway to determine a genomics-based BMD for AA in rat testes, a set of DEG related to “calcium ion binding” (Table 3) at 31 days were examined by qRT-PCR (Table 4). For the AA testes day 31 samples, the control groups were normally distributed for 6 genes (Arsi, Cacnb1, Cacng7, S100b, Tnnt2, and Trpc1). Arsi showed statistical significance (P ≤ .05) at 0.5, 1.5, 6.0, and 12.0 mg/kg using the independent t test. Cacnb1 showed statistical significance (P ≤ .05) at 1.5 and 6.0 mg/kg using the independent t test. Cacng7 showed statistical significance (P ≤ .05) at 0.5 and 12.0 mg/kg using the independent t test. S100b showed statistical significance (P ≤ .05) at 0.5, 1.5, 3.0, 6.0, and 12.0 mg/kg using the independent t test. Tnnt2 showed statistical significance (P ≤ .05) at 12.0 mg/kg using the independent t test. Trpc1 showed statistical significance (P ≤ .05) at 1.5 and 3.0 mg/kg using the independent t test. Dnase1l3 was not normally distributed in the control group and showed no statistical significance (P ≤ .05) at any dose level compared against the control group using the nonparametric Kendall correlation.
Genes Related to “Calcium Ion Binding,” as Determined Using the List of DEGs, Affected in at Least 1 Dose on Day 31 and DAVID’s Functional Annotation Analysis and BMD Modeling in the BMDExpress (32) and Data Visualization in the BMDExpress Data Viewer (34).a
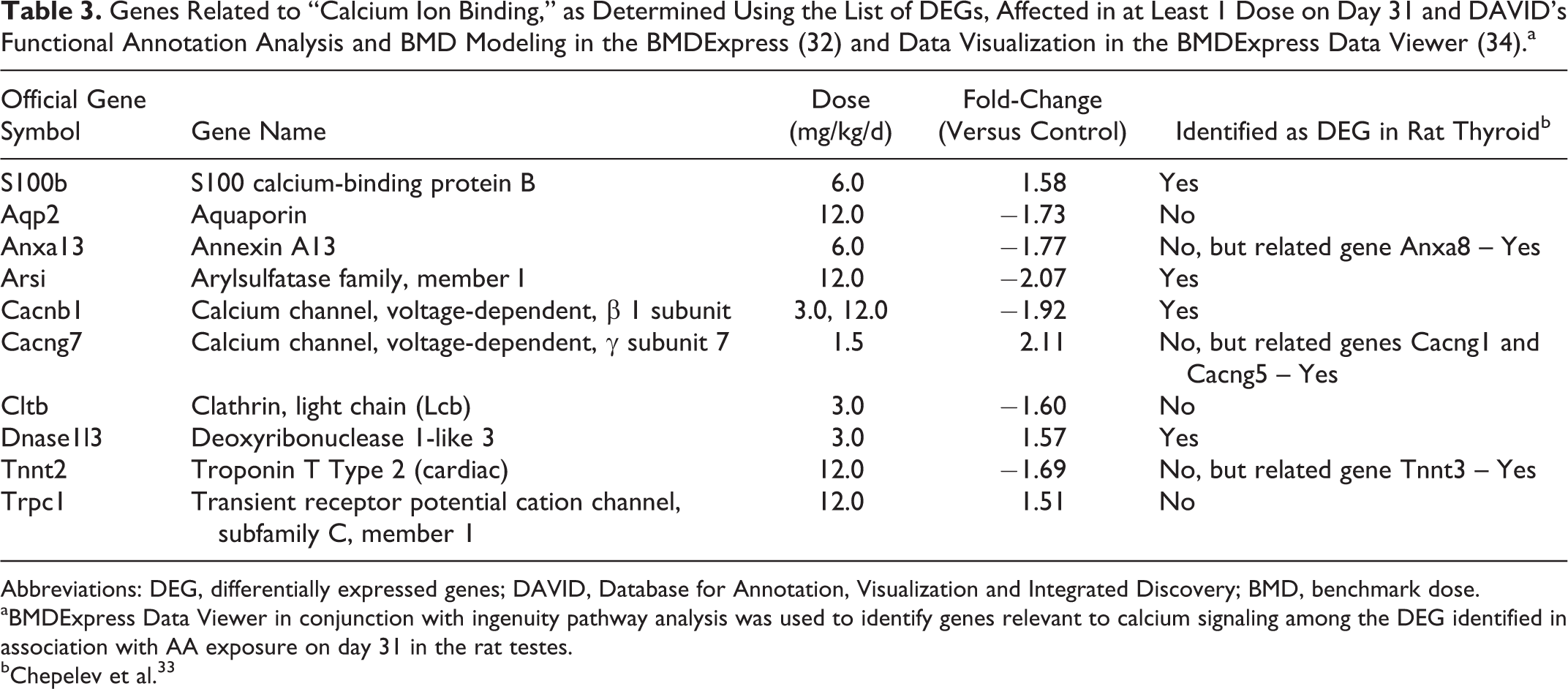
Abbreviations: DEG, differentially expressed genes; DAVID, Database for Annotation, Visualization and Integrated Discovery; BMD, benchmark dose.
aBMDExpress Data Viewer in conjunction with ingenuity pathway analysis was used to identify genes relevant to calcium signaling among the DEG identified in association with AA exposure on day 31 in the rat testes.
bChepelev et al. 33
Quantitative RT-PCR (QPCR) of Specific Target Genes Expressed as Relative Quantity (RQ) Associated With MOA of AA in Testes.a
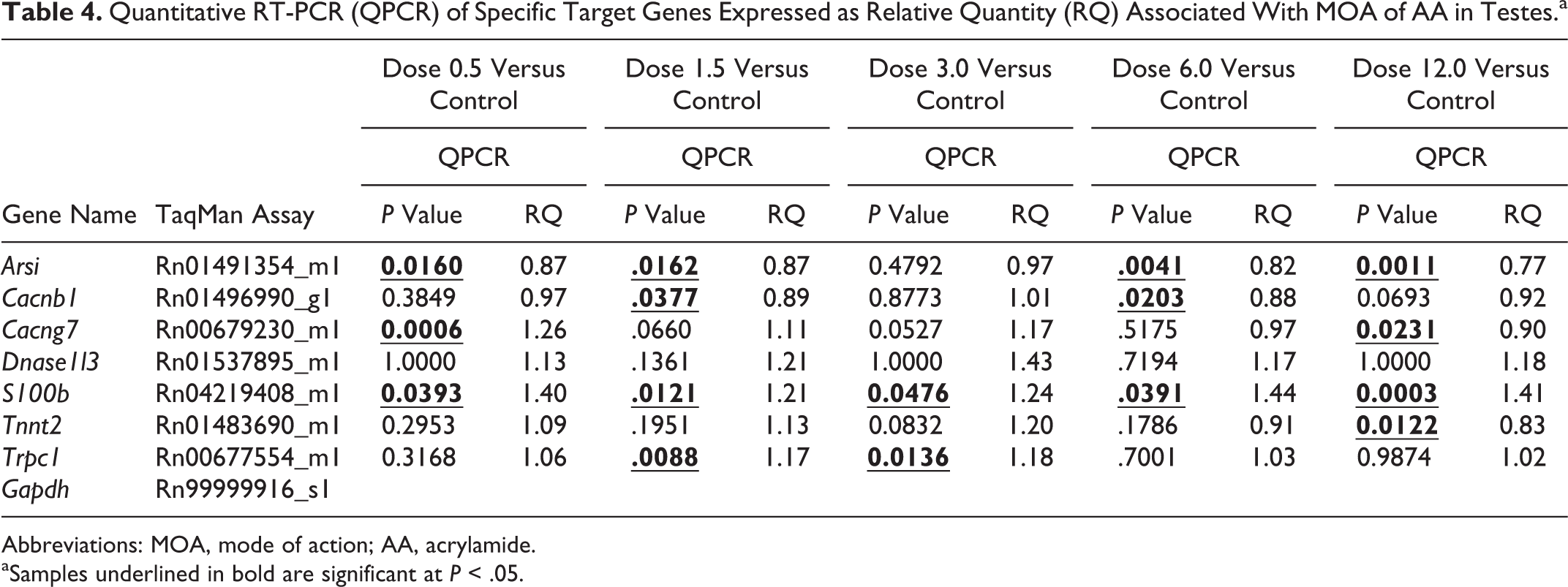
Abbreviations: MOA, mode of action; AA, acrylamide.
aSamples underlined in bold are significant at P < .05.
Benchmark Dose Analysis
BMD modeling identified only 6 pathways (with at least 2 DEG and Fisher exact test P value for a pathway <.05) as being affected by AA in the rat testes in this study. These were defined using ingenuity pathway analysis pathways (Table 5). Calcium signaling pathway was one of these pathways that could be used to determine BMD. The BMD mean for each pathway ranged from 4.84 to 9.65 mg/kg/d. The BMD lower 95% confidence limits (BMDLs) ranged from 1.8 to 6.8 mg/kg/d compared to the no-observed-adverse-effect-level of 2.0 mg/kg/d for rat reproductive toxicity. 2,4 The BMDLs determined using DEG for testes are significantly higher than the BMDL for AA using total mammary tumors in rats (0.30-0.46 mg/kg/d) or for peripheral neuropathy in rats (0.43 mg/kg/d). 4
Benchmark Dose (BMD) Modeling of Gene Expression: Cellular Signaling Pathways, Affected by AA in the Rat Testes, as Identified by BMD Modeling of All DEG (Significant in at Least 1 Dose at FDR-Adjusted P Value <.005 and Fold-Change >±1.5) on Day 31 in the BMDExpress and Examined in the BMDExpress Data Viewer.a
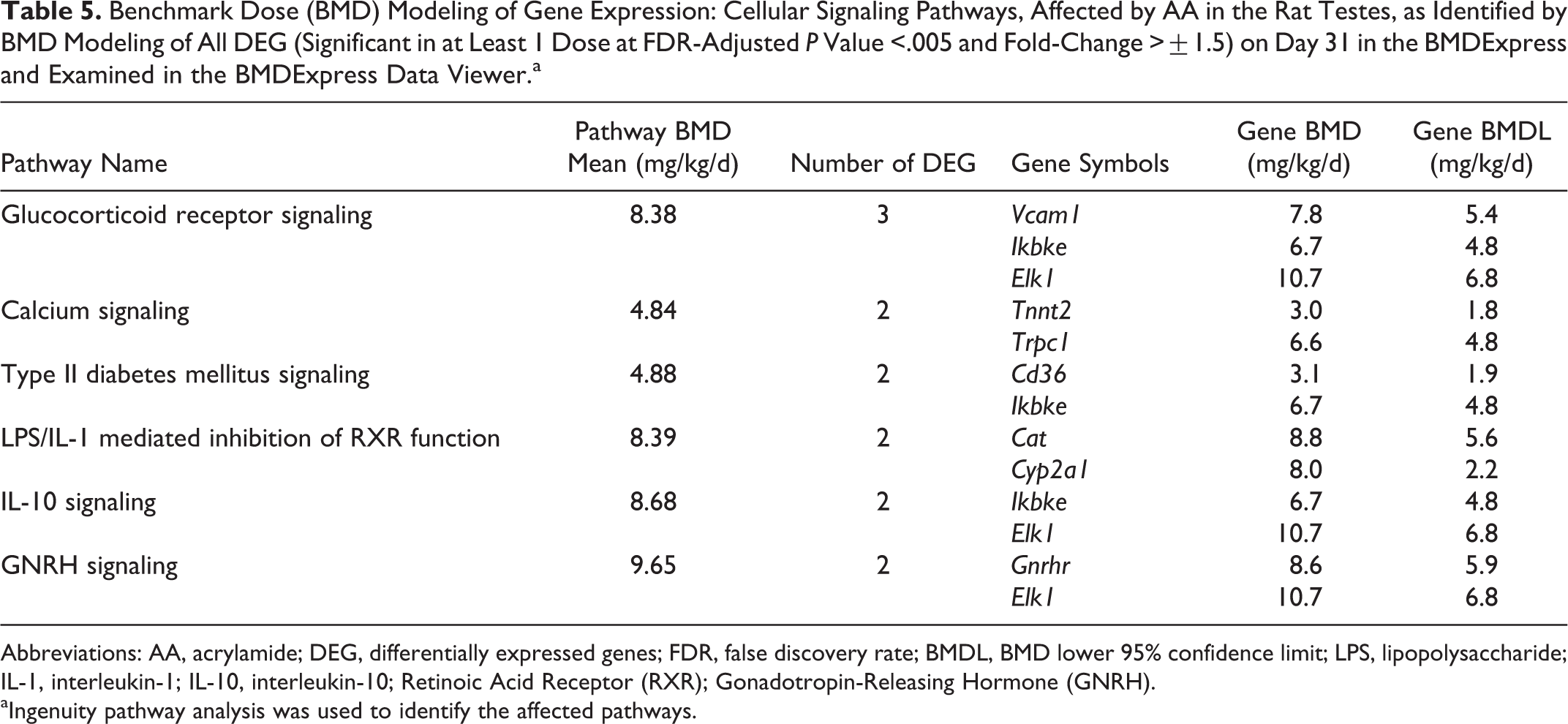
Abbreviations: AA, acrylamide; DEG, differentially expressed genes; FDR, false discovery rate; BMDL, BMD lower 95% confidence limit; LPS, lipopolysaccharide; IL-1, interleukin-1; IL-10, interleukin-10; Retinoic Acid Receptor (RXR); Gonadotropin-Releasing Hormone (GNRH).
aIngenuity pathway analysis was used to identify the affected pathways.
Discussion
The rats used in this study were part of an overall study with an assessment of genotoxicity using the micronucleus (MN) and Pig-a gene mutation assays and examining dose and time dependence of gene expression in key target tissues. In rats, there was no increase in MN and a equivocal not biologically relevant response in the Pig-a gene mutation assay. 13 These data indicate that AA is not genotoxic in the rat with respect to chromosomal aberrations as determined by the MN assay and at the exposure levels used here was not mutagenic in the Pig-a gene mutation assay. In the BigBlue™ rats, AA exposure was not mutagenic in testes. 35 In mice, however, AA is genotoxic as determined by the MN assay but not mutagenic in the Pig-a gene mutation assay. 36,37 These data indicate a species-specific genotoxic response in this study by AA in mice through the induction of chromosomal aberrations but not the induction of specific gene mutations detectable by the Pig-a assay. In rats, there is no evidence of significant genotoxicity in the bone marrow as determined by 2 well-validated genotoxicity assays conducted to meet Organization for Economic Cooperation and Development (OECD) standards for regulatory genotoxicity testing and a lack of AA mutagenicity in the testes of BigBlue™ transgenic rats. 13,35
The effect of AA on actin filaments, calcium (Ca2+) signaling, and cell proliferation was a striking observation in testes. Ca2+ is known to affect the dynamics of actin, not by directly binding to actin, but rather by modulating the activities of multiple actin regulators including protein kinase C and calmodulin-dependent kinases, both of which interact with actin affecting biological activity. 38 Previous studies have shown that AA exposure in rats affects neuronal calmodulin-dependent kinases. 7 Mechanisms leading to the altered regulation of actin filaments and Ca2+signaling genes in testes may be mediated by AA protein adducts (eg, through covalent bonds with susceptible Cys groups with cytoskeletal filaments) or through binding of AA to protamines. 4 Protamines are nuclear proteins that replace histones late in the haploid phase of spermatogenesis and are believed to be essential for sperm head condensation and DNA stabilization. Direct binding of AA to cytoskeletal proteins has also been reported previously in vitro, 23 -25 and we hypothesize that AA adducts with actin filaments can significantly alter their cellular function affecting a broad range of biological space. These data indicate that primary biological reactivity of AA in testes appears to be associated with protein binding, as direct binding of AA to testes DNA or DNA adducts mediated by the bioactivation of AA to GLY in rat testes do not appear to reach the levels needed to trigger a cellular p53 DNA damage response pathway. 39 The lack of a p53 DNA damage response in rat testes in this study is consistent with a previous microarray study in rat testes at 60 mg/kg/d of AA, 2.5 times that top dose used in this study, and with the lack mutagenicity in the testes of BigBlue™ rats administered AA in drinking water at 5 or 10 mg/kg/d for 2 months. 13,35
Ca2+ is a messenger involved in intra- and extracellular signaling networks that have a key role in spermatogenesis, cell death, and life pathways. 21,22,40 Sustained disruptions in Ca2+ signaling can have significant implications for the health and functionality of cells involved in spermatogenesis since Ca2+ signaling impacts many cellular functions including steroid signal transduction, gene expression, cytoskeletal proteins, and apoptosis. 22,41 Under resting conditions, cytosolic Ca2+ is maintained at low nanomolar concentrations by an array of pumps, buffers, and transport mechanisms. Ca2+ entry into the cytosol is rigorously controlled by intracellular compartmentalization into the endoplasmic reticulum and mitochondria. 21 In spermatogenesis, Ca2+ has a key role in signal transduction in the Leydig cell and Sertoli cellular response to testosterone. 40 A number of recent studies indicate that intracellular calcium is critical for sperm motility, capacitation, and acrosome reaction. 22 The exact roles of intracellular calcium in the regulation of the spermatogenesis are uncertain; however, calcium mobilizing channels/pumps, calmodulin, and other calcium-binding proteins are differentially expressed during mammalian spermatogenesis and in the support cells, suggesting that calcium is likely involved in the regulation of mammalian spermatogenesis. 22 The broad spectrum of toxicity in testes demonstrated by AA exposures in rats does affect both Sertoli and Leydig cells, known to have key roles in spermatogenesis and cells that respond to calcium signaling.
Ca2+ signaling also interacts with other cellular signaling systems such as reactive oxygen species (ROS). 42 Intracellular ROS at subtoxic levels also participates in cell life or death decisions, and these interactions between intracellular Ca2+ and ROS signaling pathways are thought to fine-tune the cellular life or death pathways. Therefore, dysfunction in either system can affect both and potentiate the cellular response to a stressor. In vitro exposure to AA results in mitochondrial dysfunction leading to the activation of mitochondria-driven apoptotic pathways, a cascade of aberrant redox signaling, increased in H2O2, and induction of inflammatory response genes (increased expression of inducible nitric oxide synthase). 43 In the present study, Ca2+ signaling was a primary pathway affected in testes by AA exposure, suggesting that in rat testes are in a more oxidized redox status and this can contribute to an increased level of chromosomal damage in testes of AA-exposed rats.
The lists of genes related to Ca2+ signaling were confirmed by qRT-PCR and then used to determine BMD for effects of AA exposure on Ca2+ signaling. Strikingly, out of 10 DEG related to Ca2+ signaling in this study, 4 (S100 calcium-binding protein B [S100b], arylsulfatase family, member I [Arsi], calcium channel, voltage dependent, β 1 subunit [Cacnb1], and deoxyribonuclease 1-like 3 [Dnase1l/3]) were also identified as DEG in the rat thyroid, a cancer target tissue from these same animals and their expression were altered in the same direction in both thyroid and testes. 33 Calcium signaling, but not pathways related to genotoxicity (eg, p53 DNA damage response signaling), was among the very few pathways affected by AA in this analysis of the testes transcriptome. It is of interest to also note that the BMDL calculated for gene expression in thyroid in animals from this same study was 0.68, 33 while that calculated here was 1.8 mg/kg/d suggesting that testes are not as sensitive a target tissue compared to thyroid. Taken together, these data suggest the importance of altered calcium signaling pathways with actin filament dysfunction in mediating the toxicity of AA in target tissues of rat testes.
Altered expression of calcium signaling genes and actin filaments may lead to impaired microtubule and microfilament integrity, cellular functioning, and aberrant mitosis and meiosis. Microfilaments are formed by actin proteins, are the most abundant protein within a cell, and are the key components of the cytoskeleton in eukaryotic cells. Microfilaments are highly versatile, functioning in cytokinesis and changes in cell shape and are part of the motor proteins driving cytokinesis. Microtubules have a key role in chromosome segregation and are made up of tubulin. 44 In vitro AA exposure can inhibit the microtubule motor protein kinesin, a mitotic/meiotic motor protein. 10,23 -25 Kinesins are important for proper spindle formation and are involved in sliding microtubules apart within the spindle during prometaphase and metaphase, as well as disassembly at centrosomes during anaphase. 45 AA inhibition of this same family of motor proteins in neurons is believed to be responsible for the well-known neurotoxicity of AA causing distal axonopathy, including hind-limb weakness/paralysis. 24 AA (or GLY) inhibition of kinesin may be an alternative mechanism to DNA adduction in the production of chromosomal aberrations leading to dominant lethal mutation in rat. 2,45 In this study, we found alterations in the expression of actin filament and calcium signaling genes that are impacted in testes, and we hypothesize that this may mediate dominant lethal effects in rats by compromising chromosome segregation.
Overall, the lack of measurable p53-mediated DNA damage response in testes is consistent with the notion that direct DNA binding at genotoxicity is not a primary MOA for AA-induced genotoxicity in rat testes. This is supported by a lack of significant genotoxicity observed with the MN and Pig-a assays, respectively. 13 Analysis of our toxicogenomics data alongside published toxicological studies on AA do not provide strong weight of evidence in support of MOAs centered on the direct induction of genotoxicity in rat testes. Instead, this in vivo study supports that the well-known early cytotoxic changes in response to AA exposure in the rat testes are accompanied by transcriptional alterations of the genes involved in cytoskeletal proteins and calcium signaling. This unifies and is consistent with the MOA for AA neurotoxic effects on calcium signaling cytoskeletal proteins and motor proteins as an MOA for AA testicular toxicity that could lead to dominant lethal mutation.
Footnotes
Author Contributions
Leslie Recio contributed to conception and design, contributed to acquisition, analysis, and interpretation, drafted the manuscript, and critically the revised manuscript; Marvin Friedman contributed to conception and design, contributed to interpretation, and critically revised the manuscript; Dennis Marroni contributed to conception and design, contributed to interpretation, and critically revised the manuscript; Timothy Maynor contributed to design and analysis and critically revised the manuscript; Nikolai L. Chepelev contributed to conception and design, contributed to analysis and interpretation, and critically revised the manuscript. All authors gave final approval and agree to be accountable for all aspects of work ensuring integrity and accuracy. Acknowledgments
We thank Jeff Davis, study director and the Integrated Laboratory Systems Inc Investigative Toxicology group for the in-life portion of this study. We thank Ruchir Shah and staff at Sciome for initial quality control checks of NextSeq 500 data files and identification of DEG.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was funded by SNF SAS to ILS, Inc. DM and MF are employees of SNF SAS. This work was supported by SNF SAS.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.