
Abstract
The resistance of glioblastoma to chemotherapy remains a significant clinical problem. Targeting alternative pathways such as protein prenylation is known to be effective against many cancers. Fluvastatin is a potent competitive inhibitor of 3-hydroxy-3-methylglutaryl- CoA (HMG-CoA) reductase, thereby inhibits prenylation. We demonstrate that fluvastatin alone effectively inhibits proliferation and induces apoptosis in multiple human glioblastoma cell lines. The combination index analysis shows that fluvastatin acts synergistically with common chemotherapy drugs for glioblastoma: temozolomide and irinotecan. We further show that fluvastatin acts on glioblastoma through inhibiting prenylation-dependent Ras activation. The combination of fluvastatin and low dose temozolomide resulted in remarkable inhibition of glioblastoma tumor in mice throughout the whole treatment duration without causing toxicity. Such combinatorial effects provide the basis for utilizing these FDA-approved drugs as a potential clinical approach in overcoming resistance and improving glioblastoma treatment.
Impact statement
A significant advantage of drug repositioning over traditional drug development is the rapid clinical translation of known drugs to new indications. In this work, we are the first to provide pre-clinical evidence that fluvastatin, a clinically available drug for the treatment of patients with hypercholesterolemia, displays potential to sensitize glioblastoma to chemotherapy. In addition, the underlying mechanisms of fluvastin’s action in glioblastoma are well-elaborated. Our results will be of interest not only to fundamental biological researchers but also clinical doctors.
Introduction
Glioblastoma is the most common and aggressive primary brain tumour with poor prognosis; a median survival time of 15 months after diagnosis or a 5-year survival rate of 10%. 1 Surgical resection followed by radiotherapy and chemotherapy using temozolomide represents the current standard therapy.2,3 Patients with co-deletion of complete chromosome arms 1p and 19q, methylation of the MGMT promoter or IDH1 mutations generally signified a better prognosis. 4 However, glioblastoma is inter-patient variable, intratumoral heterogeneous, and intrinsically resistance to chemotherapy. 5 This is due to the highly mutated genome of glioblastoma, which is characterized by the deregulation of key oncogenic signaling pathways involving growth, proliferation, survival, and cancer stem cell maintenance. 6 It is critical to explore novel therapeutic strategies that target these pathways. Protein farnesylation and geranylgeranylation, together referred to as prenylation, are lipid post-translational modifications that are required for proper function of many oncogenic small GTPase proteins, such as Ras superfamily proteins, through enabling their localization into cytoplasmic membrane and interactions with various signal. 7 Inhibiting prenylation thus might represent a promising therapeutic strategy to improve the treatment of glioblastoma.
The mevalonate pathway intermediates FPP (farnesyl diphosphate) and GGPP (geranylgeranyl diphosphate) that are essential enzymes for prenylation pathways. 8 Statins are HMG-CoA reductase inhibitors commonly used for the treatment of patients with hypercholesterolemia and prevention of cardiovascular diseases. 9 Due to their ability in blocking HMG-CoA reductase, an enzyme catalyzing the synthesis of mevalonate which is a critical precursor in mevalonate pathway, statins also inhibit protein prenylation and functions of many prenylation-dependent oncogenic proteins in cancer cells. 10 Fluvastatin is a member of the statin drug class and also demonstrates anti-cancer activity. Preclinical studies show that fluvastatin slows breast cancer metastasis, inhibits ovarian cancer cell proliferation and prevents non-small cell lung carcinogenesis.11–13 In this study we investigated the effect of fluvastatin on proliferation, apoptosis, protein prenylation, Ras activation, and its combinatory effect with chemotherapy drugs in glioblastoma using culturing cell system and xenograft mouse model.
Materials and methods
Cell culture, compounds, western blot and apoptosis
All human glioblastoma cell lines used in our study were authenticated via short tandem repeat profiling analysis (Procell Inc.). Cells were maintained in DMEM medium supplemented with 10% FBS and 1% penicillin/streptomycin at 37°C, 5% CO2 atmosphere. Fluvastatin, geranylgeraniol, farnesol, mevalonate, irinotecan and temozolomide were purchased from Sigma and were reconstituted in DMSO at least 1000-fold greater than experimental concentrations for in vitro assays. Western bolt was performed as described in our previous study using the antibodies from Cell Signaling. 14 Apoptosis was determined after 3 days drug treatment using Cell Death Detection ELISA (Roche). This kit measures the amount of cytoplasmic histone-associated DNA fragments (mono- and oligo nucleosomes).
Proliferation assay and combination index (CI) calculation
Cells at 5000 per sample were seeded into 96-well plate and were treated with fluvastatin for single arm analysis. After 3 days treatment, overall cell growth was assessed using BrdU (5-bromo-2'-deoxyuridine) colorimetric assay (Promega). Studies testing the combinatory effects of fluvastatin with temozolomide or irinotecan were performed using Chou-Talalay Method. Cells were treated with single drug and both drugs at increasing concentrations with 2-fold change. The constant ratio of concentration at both drugs were equipotent. After 3 days treatment, anti-proliferative effects of drug alone and combination were determined using BrdU assay. CI at any effect and dose level was automatically determined using the CalcuSyn after data entries of effects and concentrations. Quantitative definition for additive effect (CI = 1), synergism (CI < 1), and antagonism (CI > 1) in drug combinations was used. 15
Ras activation ELISA assay
Cells at 106 were seeded to 6-well plate and were treated with fluvastatin. After 24 h treatment, Ras activity was quantified using Ras activation ELISA Assay Kit (Cytoskeleton Inc.) as per manufacture’s protocol. Protein lysis were extracted from treated cells and were added to the Raf-1-RBD coated wells. Anti-Ras antibody and HRP conjugated secondary antibody. The active Ras was quantified via measuring the absorbance at 490 nm.
Overexpression of active Ras
Overexpression of active Ras was achieved by transfecting cells with HRAS Q61L plasmid containing constitutively active Ras, which is a kind gift from Dominic Esposito (Addgene plasmid # 83186). Cells at 106 were seeded to 6-well plate and were transfected with 2 µg plasmid using Lipofectamine 2000 transfection reagent (Invitrogen) as per manufacture’s protocol. Active Ras levels were examined after 48 h transfection.
Glioblastoma xenograft mouse model
All animal experiments were conducted in the veterinary facilities of Hubei University of Arts and Science in accordance to institutional rules approved by the Institutional Animal Care and Use Committee. All animal work was also conducted following NRC Guide for the Care and Use of Laboratory Animals: 8th ed. 4–6 weeks old SCID mice were purchased from Shanghai laboratory animal center and housed in a pathogen-free environment. Glioblastoma xenograft mouse model was established using the same protocol as described in our previous study. 14 107 U87 cells were implanted into mice flank. After development of palpable tumors, mice were randomized into groups for drug treatment. The mice were treated with either 20%/80% DMSO/PBS as control, low dose of temozolomide (1 mg/kg, thrice weekly), high dose of temozolomide (50 mg/kg, thrice weekly), fluvastatin (15 mg/kg, once daily), or a combination of low dose of temozolomide and fluvastatin. All drugs were resuspended in 200 ul of 20%/80% DMSO/PBS and were administrated via intraperitoneal injection using 30-gauge needle. Tumors volumes were calculated using the formula: length × width2 × 0.5236. Tumor size and body weight were monitored. After 3 weeks treatment, mice were euthanized via CO2 inhalation.
Statistical analyses
Data are expressed as mean and standard deviation (SD). Mann Whitney non-parametric test was performed for comparison of two categorical variables that are not normal or an unpaired Student’s t test for pair-wise comparisons for samples with normal assumptions. One-way analysis of variance followed by Tukey’s HSD test was conducted for multiple comparisons. p-value < 0.05 was considered as statistically significant.
Results
Fluvastatin inhibits growth and induces apoptosis in multiple glioblastoma cell lines, and is synergistic with chemotherapy drugs
To evaluate the anti-glioblastoma activity of fluvastatin, we firstly examined the anti-proliferative and pro-apoptotic capability of the fluvastatin in vitro using multiple glioblastoma cell lines. T98G, U87 and U251 are derived from different glioblastoma patients and are the most common models used in in vitro and in vivo glioblastoma research.
16
As assessed by BrdU incorporation assay, we demonstrated that fluvastatin at 1, 3 and 10 µM significantly decreased BrdU level in all tested glioblastoma cell lines in a dose-dependent manner (Figure 1(a)). Of note, treatment of fluvastatin at 10 µM led to ∼100%, ∼80% and ∼60% growth inhibition in U87, T98 G and U251 cells. In addition, fluvastatin at the same concentrations dose-dependently induces apoptosis in glioblastoma cells (Figure 1(b)), as assessed by the measurement of cytoplasmic histone-associated DNA fragments. Compared to control, fluvastatin at 10 µM increased DNA fragmentation level by 2.6-fold, 2.1-fold and 1.8-fold in U87, T98 G and U251 cells. The results obtained from proliferation and apoptosis assays on three cell lines suggest that U87 is more sensitive cell to fluvastatin treatment than T98 G and U251. Collectively, atorvastatin alone at low micromolar concentrations is effective against glioblastoma in vitro. The efficacy of fluvastatin on glioblastoma cells. Fluvastatin at 1, 3 and 10 µM significantly inhibits proliferation (a) and increases apoptosis (b) in T98 G, U87 and U251 in a dose-dependent manner. Results were obtained from at least three independent experiments and were presented as relative to control. *p < 0.05, compared to control.
We next investigated the combinatory effects of fluvastatin with chemotherapy drugs: temozolomide and irinotecan. Using Chou-Talalay Method, cells were treated with single drug and both drugs at increasing concentrations, followed by CI value calculation based on the median-effect equation.
15
As shown in Figures 2(a)–(c), the Fa-CI plot demonstrated CI<1 at in T98 G, U87 and U251 for the combination of fluvastatin and temozolomide, indicating synergism between fluvastatin and irinotecan on glioblastoma cells. Of note, synergism was also found between fluvastatin and temozolomide as shown by the Fa-CI plot (Figures 2(d)–(f)). Combination of fluvastatin with chemotherapy drugs is synergistic. Isobologram analysis of combination index (CI) values are all less than 1 in T98 G (a and d), U87 (b and e) and U251 (c and f) cells for fluvastatin and irinotecan combination, or fluvastatin and temozolomide. If CI = 1, the combination is additive; CI < 1, the combination is synergistic, and CI > 1, the combination is antagonistic.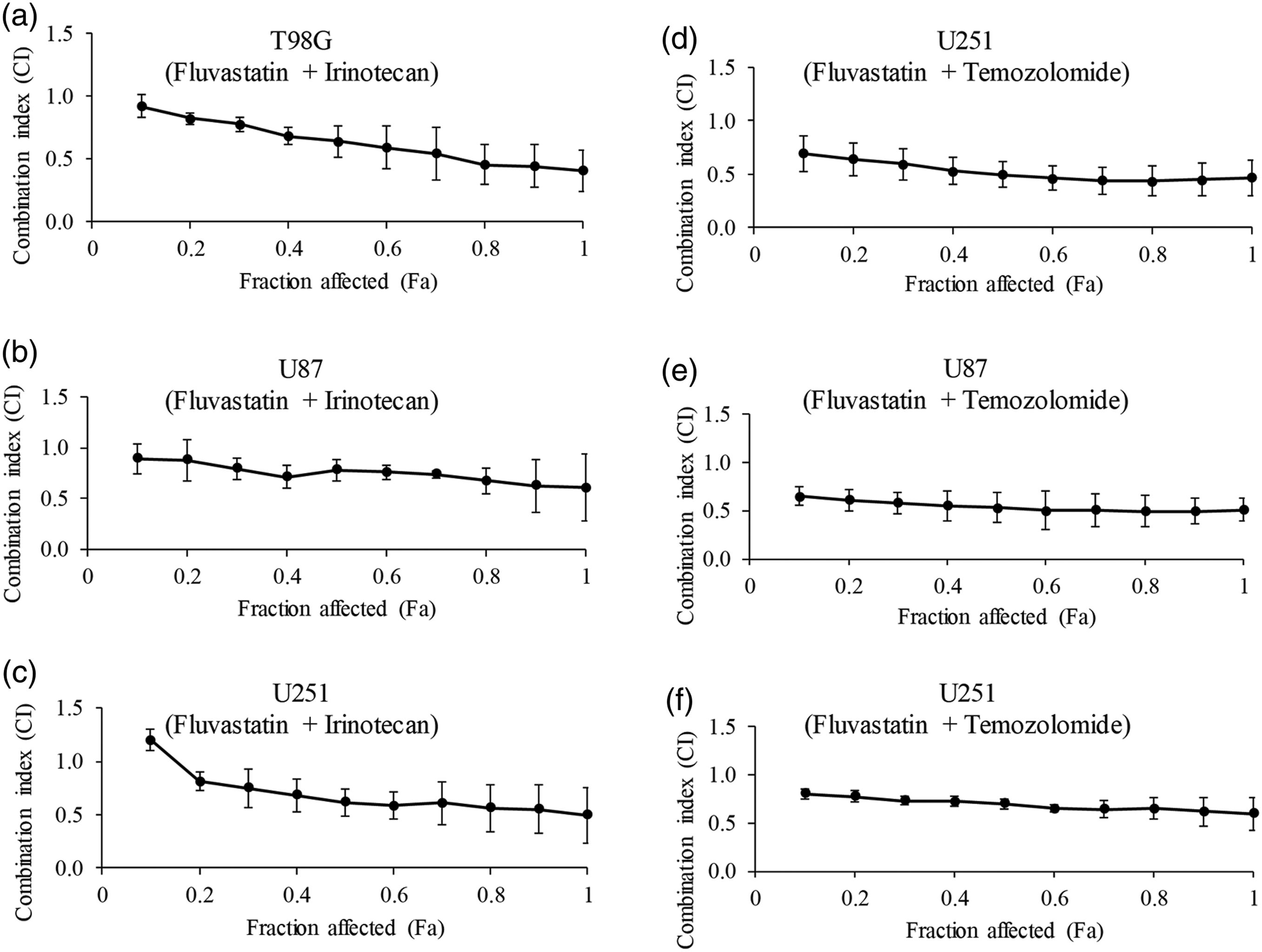
Glioblastoma cells rescued by intermediates of mevalonate pathway after fluvastatin treatment
We have previously shown that atorvastatin acts on glioblastoma via suppressing prenylation.
14
Inhibition of Ras prenylation can be monitored through immunoblotting, because its unprenylated form displays reduced mobility in SDS-PAGE compared with its prenylated form. Similar to atorvastatin, immunoblotting analysis demonstrated that fluvastatin inhibited prenylation as we observed decreased prenylated form and increased unprenylated form of Ras in U87, T98 G and U251 cells after fluvastatin treatment (Figure 3(a)). To determine which products of the mevalonate pathway have a role in the anti-glioblastoma effect of fluvastatin, we performed rescue studies by adding intermediates of prenylation pathway. Treatment with fluvastatin in the presence of mevalonate or, geranylgeraniol (GGOH) which is metabolized to geranylgeranyl pyrophosphate (GGPP) in the cells to restore geranylgeranylation but not farnesylation, reversed the anti-proliferative and pro-apoptotic effect of fluvastatin in U87, T98 G and U251 cells (Figures 3(b) and (c)). In contrast, farnesol (FOH) which is metabolized to farnesyl pyrophosphate (FPP) to exclusively restore farnesylation in the cells had no effect on fluvastatin-induced growth inhibition and apoptosis. These results confirm that prenylation inhibition attributes to fluvastatin’s action in glioblastoma. Fluvastatin acts on glioblastoma cells in a prenylation-dependent manner. (a) Representative western blot photos showing the inhibitory effect of fluvastatin on Ras prenylation. Unfarnesylated (upper band) and farnesylated (lower band) Ras were indicated. The anti-proliferative (b) and pro-apoptotic (c) effects of fluvastatin were reversed by the addition of GGOH (geranylgerniol) or MV (mevalonate) but not FOH (farnesol). 20 µM GGOH and FOH, and 50 µM MV were used. Fluvastatin and GGOH, FOH or MV were added concurrently. *p < 0.05, compared to control.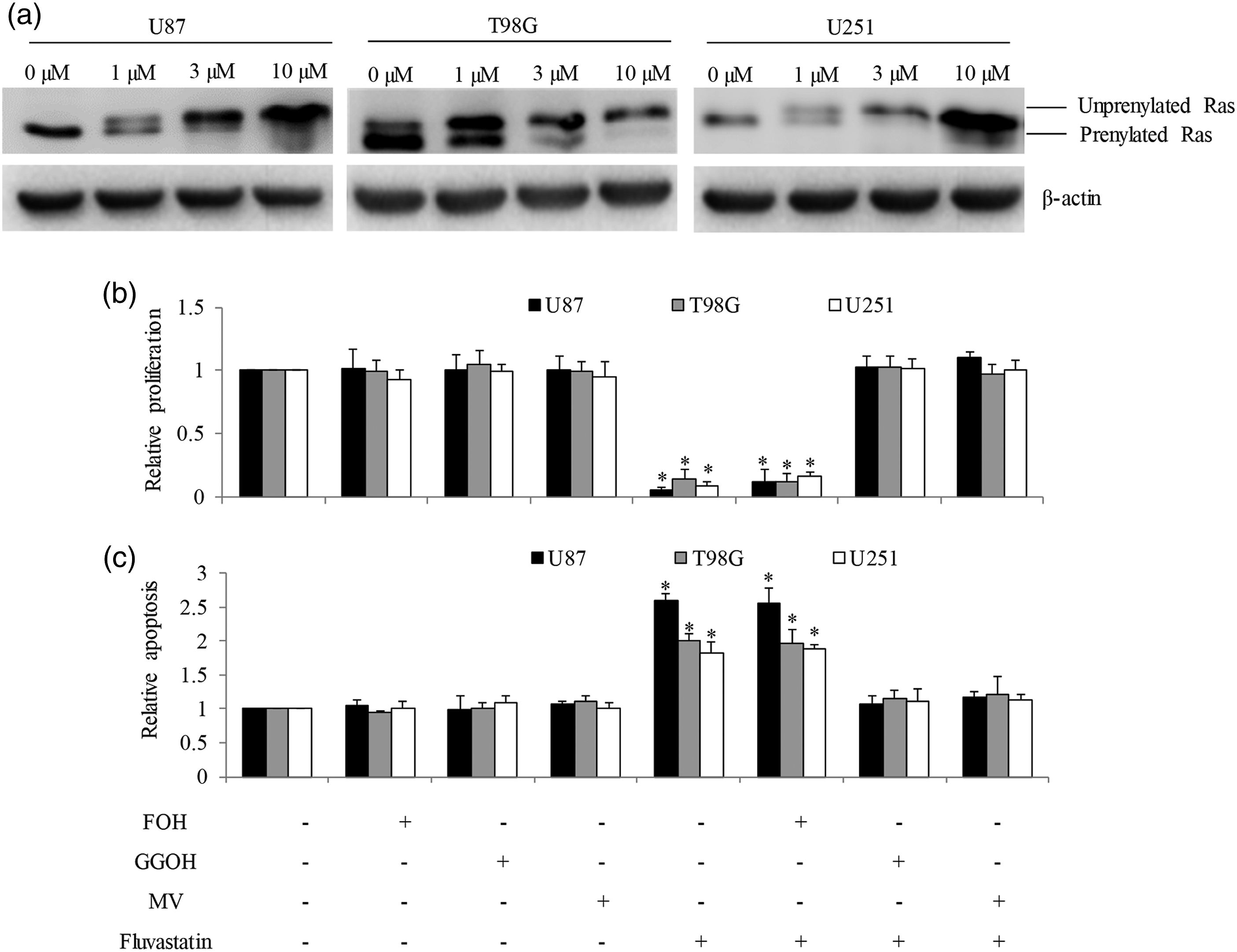
Fluvastatin inhibits Ras activation in glioblastoma cells via inhibiting prenylation
Oncogenic Ras signalling is essential for glioblastoma maintenance and its activation largely depends on prenylation.
17
To determine whether prenylation inhibition by fluvastatin leads to Ras deactivation, and whether Ras deactivation is the molecular mechanism of fluvastatin’s action in glioblastoma, we examined Ras activation in U87 cells after fluvastatin treatment and attempted to rescue fluvastatin’s effect using constitutively active Ras. As shown in Figures 4(a) and (b), fluvastatin significantly decreased active Ras level in U87 cells and this reduction was reversed by addition of mevalonate or GGOH. Overexpression of constitutively active Ras significantly rescued the decreased active Ras level, decreased proliferation and increased apoptosis after fluvastatin treatment (Figures 4(c)–(e)). Collectively, these results demonstrate that prenylation-dependent Ras deactivation by fluvastatin leads to glioblastoma cell growth arrest and death. Overexpression of constitutively active Ras rescues fluvastatin’s glioblastoma cell growth arrest and apoptosis after fluvastatin treatment. (a) Fluvastatin significantly decreases active Ras levels in U87 cells. (b) Mevalonate and geranylgerniol reversed Ras activity inhibition after fluvastatin treatment. (c) Overexpression of constitutively active Ras significantly iincreases active Ras levels in U87 cells. The inhibitory effects of fluvastatin on proliferation (d) and survival (e) were reversed in U87 cell overexpressing constitutively active Ras. *p < 0.05, compared to control.
Fluvastatin significantly enhances temozolomide’s efficacy in vivo
We finally investigated the in vivo efficacy of fluvastatin and its combination with temozolomide. We treated xenograft mouse model implanted with U87 cells with either single agent or combination, monitored tumor growth and mice body weight throughout the duration of drug treatment. We found that low dose temozolomide alone slightly and fluvastatin alone moderately decreased U87 tumor growth in mice (Figure 5(a)). Fluvastatin in combination with low dose temozolomide resulted in significantly greater efficacy compared to single drug alone in inhibiting tumor growth. The mice are well tolerated to fluvastatin, low dose temozolomide and the combination as we did not observe any body weight loss or abnormal appearance/behavior compared to control group (Figure 5(b)). Temozolomide administrated as a single drug but at a dose 50-tines higher than used in the combination treatment group was also very potent in inhibiting U87 tumor growth. However, such high dose was associated with significant body weight loss and ruffled fur-coat. These results confirm that combination of temozolomide is synergistic in inhibiting glioblastoma in vivo. The combination of fluvastatin with low dose of temozolomide is synergistic in inhibiting glioblastoma growth in vivo. (a) Fluvastatin moderately and temozolomide (low) inhibited U87 tumor growth. Co-administration of fluvastatin and low dose of temozolomide is significantly more effective than single drug alone in inhibiting U87 tumor growth. (b) A combination of fluvastatin and low dose of temozolomide did not affect mice body weight compared to control. *p < 0.05, compared to control. #p < 0.05, compared to single arm treatment.
Discussion
Novel agents and new approaches to therapy are needed to improve clinical outcomes of glioblastoma. Protein prenylation is a critical mediator in cancer and has garnered attention as therapeutic target for decades. Therapeutic intervention has focused primarily on directly targeting the prenyltransferase enzymes, FTase and GGTase, but compensatory mechanisms to single enzyme inhibition results in drug resistance. Inhibitors of mevalonate pathway enzymes which mediate prenylation and cholesterol synthesis is an alternative approach. 18 Statins represent ideal candidates for repositioning in cancer therapy because 1) a wide range of studies have suggested that tumor cells are more dependent on mevalonate pathway metabolites than their non-malignant complements; 2) statins are clinically available with known pharmacological and pharmaceutical profiling. 19 Using a platform that employed a wide range of human glioblastoma lines, we screened a library of inhibitors of mevalonate pathway including statins to identify novel drugs that could be readily introduced into glioblastoma clinical trials. We previously identified that atorvastatin can restore glioblastoma sensitivity to temozolomide. 14 In the present study, we report that fluvastatin is more potent than atorvastatin in targeting glioblastoma. The blood–brain barrier (BBB) represents a major hurdle to drug delivery to central never system. Of note, fluvastatin belongs to lipophilic statins which can easily cross the BBB in animal models. 20
Using in vitro glioblastoma cell models, we show that fluvastatin at low micromolar concentrations is effective in inhibiting proliferation and inducing apoptosis of all tested cell lines, with IC50< 3 µM. The IC50 of fluvastatin is lower than the one of atorvastatin in glioblastoma cells, 14 suggesting that fluvastatin displays higher efficacy than atorvastatin. This is supported by Jiang et al.’s work that fluvastatin, fluvastatin and pitavastatin are the three most potent statins among nine statin drugs tested including atorvastatin in all three breast cancer and glioblastoma cell lines. 21 Although we and others demonstrate the efficacy of fluvastatin is higher than many other statins in cell culturing systems, it is known that oral administration of fluvastatin has low bioavailability, extensive first-pass metabolism and short half-life. 22 We demonstrate that intraperitoneal injection rather than oral administration of fluvastatin significantly inhibits glioblastoma growth in mice, suggesting that alternative administration routes of fluvastatin is able to increase its serum concentration. Other alternative approaches to improve bioavailability of fluvastatin and enhance its anti-cancer efficacy include application of nanostructured lipid carriers, emulsomes and conjugation to activator transcription peptide.11,23–25
A significant finding of our work is that co-administration of fluvastatin with low-dose temozolomide greatly increases the potency of the latter in inhibiting glioblastoma growth in vivo, lowering the effective dose for temozolomide by ∼50-fold, with no adverse effects in mice. This is consistent with our combination index value showing that fluvastatin in combination with temozolomide or irinotecan is synergistic in inhibiting glioblastoma cell growth. Our findings are also supported by preclinical studies demonstrating the ability of fluvastatin in augmenting efficacy of not only chemotherapy drugs but also other drugs, such as valproic acid, vorinostat and aspirin, in many cancers.26-28 Metronomic treatment with carboplatin and vincristine associated with fluvastatin and thalidomide significantly increased survival of pediatric brain stem tumor patients. 29 The anti-cancer efficacy and safety of fluvastatin alone and its combination with other drugs have been shown in clinical trials. Fluvastatin (6 mg/kg/day)-celecoxib (200–800 mg /day) combination is well tolerated in children with refractory/relapsed glioma (Identifier: NCT02115074). Fluvastatin achieves measurable drug concentrations in prostatic tissue and is associated with pro-apoptotic effects on tumor cell. 30
Although the inhibitory effects of fluvastatin have been shown in various cancers, the underlying molecular mechanisms are not well understood and complex, including Sirtuin 6 upregulation, HMG-CoA reductase inhibition, Ras isoprenylation and inhibition of Akt phosphorylation.13,31,32 Similar to atorvastatin’s action in glioblastoma, 14 our mechanistic studies confirm that fluvastatin acts on glioblastoma via suppressing Ras activity, and furthermore that this is prenylation-dependent. Aberrant Ras activation due to either overexpression or mutation plays a pivotal role in regulating proliferation, survival, differentiation and signal transduction in glioblastoma.17,33,34 The ability of fluvastatin in inhibiting Ras activity makes it an attractive candidate for the treatment of not only glioblastoma but also other Ras-driven tumors.
In conclusion, we demonstrate that fluvastatin can overcome glioblastoma chemoresistance via prenylation-dependent inhibition of Ras signaling. Recent preclinical and clinical studies have demonstrated that statins can exert antitumor effect, improve clinical prognosis and significantly prolong the survival time of glioma patients. Our work adds fluvastatin to the list of statins that can be repurposed for the treatment of glioblastoma.
Footnotes
Authors’ contributions
Cheng Long and Limei Yuan performed the experiments, collated the results and wrote the manuscript; Jingwen Li and Wei Wei analysed the results and supervised the project; all revised the manuscript.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Data availability statement
Data will be made available from corresponding author upon request.
Ethics statement
The procedures with animal work were approved by the Institutional Animal Care and Use Committees of Hubei University of Arts and Science and were conducted following NRC Guide for the Care and Use of Laboratory Animals: 8th ed.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by Xiangyang Central Hospital, Hubei University of Arts and Science [Grant No. 2018-16].